苯磺顺阿曲库铵杂质及其中间体的制备方法与流程
1.本发明涉及药物合成技术领域,具体涉及苯磺顺阿曲库铵杂质及其中间体的制备方法。
背景技术:
2.苯磺顺阿曲库铵是苯磺酸阿曲库铵的10个手性异构体中的r构型全顺式异构体,如式i所示。苯磺顺阿曲库铵和苯磺酸阿曲库铵相比具有很大的优势,包括无明显组胺释放,肌松作用强,无蓄积作用,且心血管的反应小,对肝、肾等脏器功能的依赖也小,是较为理想的中时效非去极化肌松药。
[0003][0004]
单一手性构型的药物可以有比消旋药物更好的效力,也可减少服用剂量和不良反应,同时降低药物代谢的负担。苯磺顺阿曲库铵基于上述优势已成为临床肌松药的主流。
[0005]
原料药质量是药品质量控制的关键和源头,其中,杂质的研究与控制事关药品的临床安全性,因而成为原料药质量控制的关键环节之一。苯磺顺阿曲库铵结构中有4个手性中心,共10个手性异构体,理论上讲除r全顺式构型(即苯磺顺阿曲库铵)外,另外9个都是潜在的杂质。
[0006]
欧洲药典10.0版列出了需要控制的苯磺顺阿曲库铵杂质共22个,其中属于手性异构体的杂质有6个,为g、h、s、t、u、v。其中g、h属于1r-1'r的构型,s、t、u、v这四个属于1r,1's的构型。(未被列出的3个杂质结构均为1s,1's构型,从工艺路线上分析,生成这三种结构的概率低到可以忽略不计)
[0007]
其中杂质s为1r-顺-1's-顺式苯磺酸阿曲库铵,如式ii所示
[0008][0009]
杂质v为1r-反-1's-顺式苯磺酸阿曲库铵,如式iii所示
[0010][0011]
现有进行苯磺顺阿曲库铵质量控制时所采用的杂质都是采用进口标准品,价格异常昂贵,并且每批次都要使用,因而在一定程度上大大提升了成本,如果能够开发出上述杂质的适宜制备方法,对于大生产中经济效益的提升势必大有帮助,还能进一步提升苯磺顺阿曲库铵的质量控制。
技术实现要素:
[0012]
针对现有技术存在的上述问题,本发明提供苯磺顺阿曲库铵杂质及其中间体的制备方法。本发明的技术方案为:
[0013]
第一方面,本发明提供苯磺顺阿曲库铵杂质s和杂质v中间体的制备方法,是以四氢罂粟碱盐酸盐为起始原料,游离后用n-乙酰基-l亮氨酸成盐拆分,得到r-四氢罂粟碱和s-四氢罂粟碱;r-四氢罂粟碱和s-四氢罂粟碱再分别与丙烯酸叔丁酯缩合,再与苯磺酸甲酯进一步形成季铵盐,结晶后得到三个中间体:式iv显示的为s-顺式酯化合物、式vi显示的为r-顺式酯化合物和式vii显示的为r-反式酯化合物;
[0014][0015]
具体反应方程式为:
[0016][0017]
以及,
[0018][0019]
进一步地,上述制备方法包括以下步骤:
[0020]
(a)将四氢罂粟碱盐酸盐混悬于甲苯与水中,加足量氨水搅拌游离,经洗涤、干燥后浓缩;然后与n-乙酰-l-亮氨酸成盐析晶,过滤得到s-四氢罂粟碱-n-乙酰-l-亮氨酸盐粗品,滤液蒸干得到r-四氢罂粟碱-n-乙酰-l-亮氨酸盐粗品;
[0021]
(b)将s-四氢罂粟碱-n-乙酰-l-亮氨酸盐粗品和r-四氢罂粟碱-n-乙酰-l-亮氨酸盐粗品分别重结晶,得到s-四氢罂粟碱-n-乙酰-l-亮氨酸盐精制品和r-四氢罂粟碱-n-乙酰-l-亮氨酸盐精制品;
[0022]
(c)将s-四氢罂粟碱-n-乙酰-l-亮氨酸盐混悬于甲苯与水中,加足量氨水搅拌游
离,经洗涤、干燥后浓缩;然后先与丙烯酸叔丁酯进行加成缩合反应,反应结束后将产物与草酸成草酸盐后提纯;草酸盐游离后再与苯磺酸甲酯生成s-季铵盐混旋体,经过三次精制,得到s-顺式酯化合物;
[0023]
(e)r-四氢罂粟碱-n-乙酰-l-亮氨酸盐的反应过程同步骤(c),得到r-季铵盐混旋体,经精制得到r-顺式酯化合物和r-反式酯化合物。
[0024]
进一步地,所述步骤(a)中四氢罂粟碱盐酸盐与n-乙酰-l-亮氨酸的摩尔比为1:(1~1.1)。
[0025]
进一步地,所述步骤(a)中浓缩物与n-乙酰-l-亮氨酸成盐析晶的控制条件包括:成盐温度为80
±
2℃,成盐析晶溶剂为无水乙醇,用量为8~12ml/g浓缩物,析晶温度不高于15℃。
[0026]
进一步地,所述步骤(b)中s-四氢罂粟碱-n-乙酰-l-亮氨酸盐粗品重结晶的过程包括:将s-四氢罂粟碱-n-乙酰-l-亮氨酸盐粗品于80
±
2℃溶于8~10ml/g粗品的无水乙醇中,然后将温度降至45℃以下搅拌30~40min,过滤得到s-四氢罂粟碱-n-乙酰-l-亮氨酸盐精制品。
[0027]
进一步地,所述步骤(b)中r-四氢罂粟碱-n-乙酰-l-亮氨酸盐粗品重结晶的过程包括:将r-四氢罂粟碱-n-乙酰-l-亮氨酸盐粗品用丙酮和水混合溶剂加热回流溶解,梯度降温至25℃以下搅拌3~4h,过滤得到r-四氢罂粟碱-n-乙酰-l-亮氨酸盐精制品。
[0028]
进一步地,所述丙酮和水混合溶剂中丙酮用量为26~32ml/g粗品,优选为30ml/g粗品;水用量为0.1~1ml/g粗品,优选为0.3ml/g粗品。
[0029]
优选地,所述梯度降温速度为10℃/h。
[0030]
进一步地,所述步骤(c)中加成缩合反应的控制条件包括:反应溶剂为甲苯,催化剂为冰乙酸,反应温度为60
±
3℃,反应时间为5~7h。
[0031]
进一步地,所述步骤(c)中三次精制的过程包括:
[0032]
1)将s-季铵盐混旋体加入二氯甲烷于32℃以上溶解,再加入甲基叔丁基醚,并于29
±
2℃搅拌析晶,有固体析出后继续保温析晶4h
±
10min,过滤并用甲基叔丁基醚洗涤滤饼,该滤饼为s-反式酯化合物;
[0033]
2)将步骤1)的滤液和洗涤液合并后浓缩至干,加二氯甲烷溶解,再加入乙酸乙酯于29
±
1℃搅拌析晶,有固体析出后保温析晶3h
±
10min,过滤后得到s-顺式酯粗品;
[0034]
3)将s-顺式酯粗品加入二氯甲烷于32℃以上溶解,再加入乙酸乙酯,于29
±
1℃搅拌析晶,有固体析出后继续保温析晶3h
±
10min,过滤出固体并用乙酸乙酯洗涤,烘干后得到s-顺式酯,即式iv化合物。
[0035]
进一步地,所述步骤(e)中精制的过程包括:
[0036]
(1)将r-季铵盐混旋体加入二氯甲烷于32℃以上溶解,再加入甲基叔丁基醚,并于29
±
2℃搅拌析晶,有固体析出后继续保温析晶4h
±
10min,过滤并用甲基叔丁基醚洗涤滤饼,分别收集滤饼,以及滤液与洗涤液的混合液;
[0037]
(2)将滤饼于37
±
1℃溶于二氯甲烷中,加入甲基叔丁基醚,并降至22
±
1℃搅拌20
±
10min,过滤出沉淀,干燥后得到r-反式酯,即式vii化合物;
[0038]
(3)将滤液与洗涤液的混合液浓缩至干,加二氯甲烷溶解,再加入乙酸乙酯于29
±
1℃搅拌析晶,有固体析出后保温析晶3h
±
10min,过滤后得到r-顺式酯粗品;
[0039]
(4)将r-顺式酯粗品加入二氯甲烷于32℃以上溶解,再加入乙酸乙酯,于29
±
1℃搅拌析晶,有固体析出后继续保温析晶3h
±
10min,过滤出固体并用乙酸乙酯洗涤,烘干后得到r-顺式酯,即式vi化合物。
[0040]
第二方面,本发明提供苯磺顺阿曲库铵杂质s和杂质v的制备方法,是将s-顺式酯化合物水解后,先与1,5-戊二醇形成单酯化产物,单酯化产物再分别与r-顺式酯化合物和r-反式酯化合物的水解物酯化,进而获得杂质s和杂质v,杂质s和杂质v的结构式为:
[0041][0042]
具体反应方程式为:
[0043][0044]
进一步地,上述制备方法包括以下步骤:
[0045]
i)将s-顺式酯在苯磺酸水溶液中于40
±
3℃水解,水解产物与1,5-戊二醇反应,制得s-顺式酯单酯化化合物;
[0046]
ii)将r-顺式酯化合物在苯磺酸水溶液中于40
±
3℃水解,然后加入s-顺式酯单酯化化合物在甲苯中搅拌,于25~35℃减压回流24小时
±
10min,期间不间断分出生成的水,反应结束后洗涤、干燥,滴入甲基叔丁基醚中析晶得到杂质s的粗品,该粗品经乙醚重结晶后即得苯磺顺阿曲库铵杂质s纯品;
[0047]
iii)将r-反式酯化合物在苯磺酸水溶液中于40
±
3℃水解,然后加入s-顺式酯单酯化化合物在甲苯中搅拌,于25~35℃减压回流24小时
±
10min,期间不间断分出生成的水,反应结束后洗涤、干燥,滴入乙醚中析晶得到杂质v的粗品,该粗品经甲基叔丁基醚重结晶后即得苯磺顺阿曲库铵杂质v纯品。
[0048]
本发明采用的技术方案合成工艺安全可靠,反应重现性好,成为一种高效制备苯磺顺阿曲库铵杂质s和v的方法。应用该方法制备的化合物纯度较高,可以作为苯磺顺阿曲库铵质量控制的杂质对照品。
附图说明
[0049]
此处所说明的附图用来提供对本发明的进一步理解,构成本发明的一部分,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
[0050]
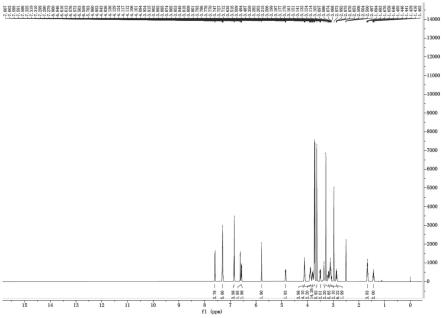
图1是本发明实施例4获得的式vi化合物的1h核磁谱图。
[0051]
图2是本发明实施例5获得的式vii化合物的1h核磁谱图。
[0052]
图3是本发明实施例7获得的式ii化合物(杂质s)uplc色谱图。
[0053]
图4是本发明实施例7获得的式ii化合物(杂质s)1h核磁谱图。
[0054]
图5是本发明实施例8获得的式iii化合物(杂质v)uplc色谱图。
[0055]
图6是本发明实施例8获得的式iii化合物(杂质v)1h核磁谱图。
具体实施方式
[0056]
在本发明的描述中,需要说明的是,实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。以下实施例的说明只是用于帮助理解本发明的方法及其核心思想,并不用以限制本发明,应当指出,对于本领域的技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
[0057]
本发明具体实施例采用的苯磺顺阿曲库铵杂质及其中间体合成路线具体如下:
[0058][0059]
;以及
[0060][0061]
在本发明的实施例和对比例中,测定纯度的方法为:
[0062]
稀释剂:乙腈:磷酸二氢钾溶液(称取6.8g磷酸二氢钾,用1l水溶解,磷酸调节其ph至3.1)=20:80。
[0063]
取本品,加稀释剂溶解并稀释制成每1ml中含0.6mg的溶液作为供试品溶液。
[0064]
色谱柱:acquity uplc beh c18 1.7μm 2.1
×
100mm;
[0065]
柱温:35℃;
[0066]
流动相:流动相a:甲醇:乙腈:磷酸二氢钾溶液(6.8g/l)(v:v:v)=5:15:80;
[0067]
流动相b:甲醇:乙腈(v:v)=30:70;
[0068]
流速:0.5ml/min;
[0069]
检测波长:280nm;
[0070]
进样量:2μl;
[0071]
样品盘温度:6℃
±
2℃;
[0072]
照以上色谱条件试验,面积归一化法计算。
[0073]
下面结合附图和具体的实施例对本发明做进一步详细说明,所述是对本发明的解释而不是限定。
[0074]
实施例1
[0075]
s-四氢罂粟碱-n-乙酰-l-亮氨酸盐的制备
[0076]
向50l玻璃反应釜中加入纯化水9.6kg,开启搅拌,投入四氢罂粟碱盐酸盐3.5kg,搅拌10分钟,投入甲苯6.2kg,搅拌20分钟,用氨水3.0kg调节ph≥9,搅拌15~20分钟,物料溶清后静置30分钟,分层;水层转入另一50l玻璃反应釜中,加入甲苯3.5kg,搅拌15分钟,静置30分钟分层,合并甲苯层,加入纯化水6.0kg,搅拌15分钟,静置30分钟分层,水层报废处理,有机层中加入无水硫酸钠0.6kg,常温搅拌1小时,过滤,滤液转移至100l玻璃釜内,将反应釜夹套温度设为60
±
5℃、真空度≥0.085mpa减压蒸馏至无连续液滴馏出,向釜内加入无水乙醇39.7kg,控制夹套温度55
±
5℃搅拌使固体全部溶解,溶清后加入n-乙酰-l-亮氨酸1.63kg,升温至80
±
4℃回流状态下保温反应1.5小时,将釜内温度降至15
±
3℃,待有明显固体析出时开始计时,保温4小时,离心甩滤,滤饼与滤液a分别处理。
[0077]
滤饼烘干后得到类白色固体2.2kg,液相检测证明其中s构型占81.2%。用17.5kg乙醇与75-80℃搅拌溶解,随后自然降温,降至45℃后搅拌0.5小时,减压抽滤,用3.0kg无水乙醇淋洗滤饼,滤饼于45℃真空干燥12小时,得到1.4kg s-四氢罂粟碱-n-乙酰-l-亮氨酸盐,收率29.3%,化学纯度100%,光学纯度99.9%。
[0078]
实施例2
[0079]
r-四氢罂粟碱-n-乙酰-l-亮氨酸盐的制备
[0080]
将实施例1中的滤液a(液相显示其中r构型为79.2%)转移至100l玻璃釜内,夹套温度50
±
5℃、真空度≥0.085mpa减压蒸馏至无连续馏分馏出,得到浓缩物。加入丙酮58.9kg,控制夹套温度60
±
5℃升温至回流状态,搅拌使固体大部分溶解后,加入纯化水0.6kg,搅拌至溶清;溶清后,逐渐降至25℃析晶,待明显析出固体后计时,23~26℃保温3.5小时,减压抽滤,滤饼用10℃以下的丙酮0.8kg淋洗,烘干后得到1.25kg白色固体,为r-四氢罂粟碱-n-乙酰-l-亮氨酸盐,收率26.2%。取样检测,化学纯度100%,光学纯度99.9%。
[0081]
实施例3
[0082]
式iv化合物(s-顺式酯)的制备
[0083]
向100l玻璃釜中加入纯化水11.2kg、甲苯11.2kg、s-四氢罂粟碱-n-乙酰-l-亮氨酸盐1.25kg,室温搅拌20~25分钟,投入浓氨水0.96kg调ph≥9,搅拌10~20分钟使物料溶清;溶清后静置15~20分钟,分层,水层转入50l玻璃反应釜中,水层中加入甲苯3.75kg,搅
拌15分钟,静置30分钟分层;水层报废处理,有机层合并至100l玻璃釜中,加入纯化水3.75kg,搅拌15分钟,静置20分钟,水层报废处理,有机层加入无水硫酸钠0.63kg,常温搅拌1小时,过滤,滤饼用甲苯0.5kg淋洗;滤液转移至50l玻璃釜中,控制夹套温度60
±
5℃、真空度≥0.08mpa减压蒸除溶剂,至无连续液滴馏出;加入甲苯0.63kg,丙烯酸叔丁酯0.77kg,冰乙酸0.08kg,设置夹套温度60
±
3℃保温反应6小时,控制夹套温度55~60℃,真空度≥0.85mpa下减压浓缩至无连续液滴滴下。
[0084]
称取草酸0.48kg溶于丙酮5.0kg中,再将乙酸乙酯2.0kg加入上述丙酮溶液中备用;向上步浓缩液中加入乙酸乙酯2.0kg溶解,得缩合物乙酸乙酯溶液转入草酸溶液中,开启搅拌,有固体析出,充分搅拌60分钟后降温至10
±
3℃搅拌析晶6小时。过滤,滤饼用丙酮0.63kg充分淋洗,滤饼烘干后得s-缩合物草酸盐约1.17kg。
[0085]
向100l不锈钢反应釜中投入纯化水10.5kg、s-缩合物草酸盐1.17kg、再加入甲苯10.5kg、搅拌下加入氨水0.80kg,调节至ph≥9.0,搅拌30分钟,静置30分钟,分层,水层转移至50l玻璃反应釜中,加入甲苯3.5kg,搅拌10~20分钟,静置30分钟,分层,水层弃去,有机层合并至100l不锈钢反应釜中,加入纯化水3.5kg,搅拌10~20分钟,静置30分钟,分层,水层弃去,有机层加入无水硫酸钠0.55kg,常温搅拌10~20min,过滤,滤液转移至50l不锈钢反应釜中,反应釜夹套温度设为55~60℃,真空度≥0.85mpa下减压浓缩至无连续液滴滴下;向浓缩物中投入苯磺酸甲酯0.64kg、乙腈0.35kg,控制釜内温度25~30℃保温反应15小时;称取乙酸乙酯1.22kg投入反应釜内,搅拌溶解,搅拌下加入甲基叔丁基醚2.63kg,25
±
3℃搅拌析晶6小时;过滤,滤饼用甲基叔丁基醚0.4kg淋洗,烘干后得s-构型季铵盐混旋体1.12kg,uplc检测其中s-顺式占75.0%,s-反式占24.3%。
[0086]
向50l玻璃釜中投入s-构型季铵盐混旋体1.12kg、二氯甲烷7.05kg,升温至32℃以上,搅拌溶解,称取甲基叔丁基醚6.23kg,搅拌下加入二氯甲烷溶液中,控制釜内温度29
±
2℃搅拌析晶4小时以上(明显析出固体开始计时);过滤,滤饼用甲基叔丁基醚0.44kg洗涤,滤液转移回50l玻璃釜内,控制夹套温度30~40℃减压浓缩,浓缩至干(固体)时加入二氯甲烷1.65kg,釜内温度升至32℃以上溶解,再加入乙酸乙酯3.30kg,控制釜内温度29
±
1℃待有明显固体析出时开始计时,保温析晶3h,过滤,滤饼用乙酸乙酯0.37kg,滤饼烘干后,得0.64kg s-顺式酯粗品,液相显示纯度为97.2%。
[0087]
向10l玻璃釜中投入二氯甲烷1.14kg、s-顺式酯粗品0.64kg,釜内温度升至32℃以上溶解,再加入乙酸乙酯2.28kg,控制釜内温度29
±
1℃待有明显固体析出时开始计时,保温析晶3h,过滤,滤饼用乙酸乙酯0.26kg洗涤,滤饼于35℃真空干燥10小时,得式iv化合物(s-顺式酯)0.46kg,收率29.5%,纯度99.3%。
[0088]
实施例4
[0089]
式vi化合物(r-顺式酯)的制备
[0090]
向100l玻璃釜中加入纯化水11.2kg、甲苯11.2kg、r-四氢罂粟碱-n-乙酰-l-亮氨酸盐1.25kg,室温搅拌20~25分钟,投入浓氨水0.96kg调ph≥9,搅拌10~20分钟使物料溶清;溶清后静置15~20分钟,分层,水层转入50l玻璃反应釜中,水层中加入甲苯3.75kg,搅拌15分钟,静置30分钟分层;水层报废处理,有机层合并至100l玻璃釜中,加入纯化水3.75kg,搅拌15分钟,静置20分钟,水层报废处理,有机层加入无水硫酸钠0.63kg,常温搅拌1小时,过滤,滤饼用甲苯0.5kg淋洗;滤液转移至50l玻璃釜中,控制夹套温度60
±
5℃、真空
度≥0.08mpa减压蒸除溶剂,至无连续液滴馏出;加入甲苯0.63kg,丙烯酸叔丁酯0.77kg,冰乙酸0.08kg,设置夹套温度60
±
3℃保温反应6小时,控制夹套温度55~60℃,真空度≥0.85mpa下减压浓缩至无连续液滴滴下。
[0091]
称取草酸0.48kg溶于丙酮5.0kg中,再将乙酸乙酯2.0kg加入上述丙酮溶液中备用;向上步浓缩液中加入乙酸乙酯2.0kg溶解,得缩合物乙酸乙酯溶液转入草酸溶液中,开启搅拌,有固体析出,充分搅拌60分钟后降温至10
±
3℃搅拌析晶6小时。过滤,滤饼用丙酮0.63kg充分淋洗,滤饼烘干后得r-缩合物草酸盐约1.17kg。
[0092]
向100l不锈钢反应釜中投入纯化水10.5kg、r-缩合物草酸盐1.17kg、再加入甲苯10.5kg、搅拌下加入氨水0.80kg,调节至ph≥9.0,搅拌30分钟,静置30分钟,分层,水层转移至50l玻璃反应釜中,加入甲苯3.5kg,搅拌10~20分钟,静置30分钟,分层,水层弃去,有机层合并至100l不锈钢反应釜中,加入纯化水3.5kg,搅拌10~20分钟,静置30分钟,分层,水层弃去,有机层加入无水硫酸钠0.55kg,常温搅拌10~20min,过滤,滤液转移至50l不锈钢反应釜中,反应釜夹套温度设为55~60℃,真空度≥0.85mpa下减压浓缩至无连续液滴滴下;向浓缩物中投入苯磺酸甲酯0.64kg、乙腈0.35kg,控制釜内温度25~30℃保温反应15小时;称取乙酸乙酯1.22kg投入反应釜内,搅拌溶解,搅拌下加入甲基叔丁基醚2.63kg,25
±
3℃搅拌析晶6小时;过滤,滤饼用甲基叔丁基醚0.4kg淋洗,烘干后得r-构型季铵盐混旋体1.12kg,uplc检测其中r-顺式占75.0%,r-反式占24.3%。
[0093]
向50l玻璃釜中投入r-构型季铵盐混旋体1.12kg、二氯甲烷7.05kg,升温至32℃以上,搅拌溶解,称取甲基叔丁基醚6.23kg,搅拌下加入二氯甲烷溶液中,控制釜内温度29
±
2℃搅拌析晶4小时以上(明显析出固体开始计时);过滤,滤饼用甲基叔丁基醚0.44kg洗涤。滤饼标计为b,用于下文式vii化合物(r-反式酯)的制备。
[0094]
滤液转移回50l玻璃釜内,控制夹套温度30~40℃减压浓缩,浓缩至干(固体)时加入二氯甲烷1.65kg,釜内温度升至32℃以上溶解,再加入乙酸乙酯3.30kg,控制釜内温度29
±
1℃待有明显固体析出时开始计时,保温析晶3h,过滤,滤饼用乙酸乙酯0.37kg,滤饼烘干后,得0.64kg r-顺式酯粗品,液相显示纯度为97.2%。
[0095]
向10l玻璃釜中投入二氯甲烷1.14kg、r-顺式酯粗品0.64kg,釜内温度升至32℃以上溶解,再加入乙酸乙酯2.28kg,控制釜内温度29
±
1℃待有明显固体析出时开始计时,保温析晶3h,过滤,滤饼用乙酸乙酯0.26kg洗涤,滤饼于35℃真空干燥10小时,得式iv化合物(r-顺式酯)0.46kg,收率29.5%,uplc纯度99.3%。核磁谱图如图1所示:1h-nmr(600m,dmso-d6)δ=1.38(s,9h),2.86-2.89(m,1h),2.94-2.97(m,2h),3.06-3.09(m,2h),3.25(s,3h),3.29(s,3h),3.48-3.52(m,2h),3.56-3.58(m,1h),3.65-3.68(m,4h),3.71(s,3h),3.73(s,3h),3.86-3.88(m,1h),4.71(dd,1h,j=10.8,4.2hz),5.68(s,1h),6.53(dd,1h,j=8.4,1.8hz),6.70(d,1h,j=1.8hz),6.84(m,2h),7.29-7.32(m,3h),7.60-7.62(m,2h)。
[0096]
实施例5
[0097]
式vii化合物(r-反式酯)的制备
[0098]
将实施例4中的滤饼b于35℃下真空干燥10小时,得到0.20kg白色固体,uplc检测判断其中含r-反式酯97.7%,将该固体于37℃溶于0.8l二氯甲烷中,将0.8l甲基叔丁基醚加入该溶液中,降至22℃后再搅拌20分钟,减压抽滤,用0.20l甲基叔丁基醚淋洗滤饼,滤饼于35℃真空干燥10小时,得到0.19kg式vii化合物(r-反式酯),为白色固体,收率12.2%(以
r-四氢罂粟碱-n-乙酰-l-亮氨酸盐投料量计)。uplc纯度99.9%。核磁谱图如图2所示:1h-nmr(600m,dmso-d6)δ=1.38(s,9h),2.85-2.89(m,1h),2.93-2.96(m,2h),3.06-3.08(m,2h),3.25(s,3h),3.28(s,3h),3.48-3.51(m,2h),3.55-3.58(m,1h),3.65-3.69(m,4h),3.71(s,3h),3.73(s,3h),3.83-3.89(m,1h),4.70(dd,1h,j=10.8,4.2hz),5.68(s,1h),6.53(dd,1h,j=8.4,1.8hz),6.69(d,1h,j=1.8hz),6.84(m,2h),7.29-7.32(m,3h),7.60-7.61(m,2h)。
[0099]
实施例6
[0100]
式v化合物(s-顺式酯单酯化化合物)的制备
[0101]
向三口瓶中加入70%苯磺酸水溶液8.0g、纯化水3.5g和式iv化合物(s-顺式酯)5.0g(1.0eq),控制釜内温度40
±
3℃搅拌约60分钟,至固体完全溶解,旋蒸蒸除其中水分后,加20ml二氯甲烷溶解。将该溶液滴入含有1,5-戊二醇16.1g(20eq),caso4(17.0g)的60ml二氯甲烷悬浊液中,环境温度下搅拌18小时,减压抽滤,滤液再加30ml二氯甲烷稀释,用纯化水洗3
×
40ml,经无水硫酸镁干燥后,旋蒸至干,得到白色固体4.6g,收率87.9%。
[0102]
实施例7
[0103]
式ii化合物(杂质s,1r-顺-1's-顺式苯磺酸阿曲库铵)的制备
[0104]
向三口瓶中加入70%苯磺酸水溶液8.0g、纯化水3.5g和式vi化合物(r-顺式酯)5.0g(1.0eq),控制釜内温度40
±
3℃搅拌约60分钟,至固体完全溶解,旋蒸蒸除其中水分后,加100ml甲苯和式v化合物5.23g(1.0eq)。装置分水器与回流冷凝管后(分水器中有适量4a分子筛用于吸水),内温控制在25-35℃减压回流反应24小时,静置后分出上层并舍弃,下层用90ml二氯甲烷溶解,用纯化水洗2
×
65ml,经无水硫酸镁干燥后,滴入900ml甲基叔丁基醚中析晶,减压抽滤,得到白色固体6.5g。该粗品经制备液相分离纯化,旋蒸浓缩至约30ml,滴入400ml乙醚中析晶,减压抽滤,将滤饼于35℃下真空干燥6小时,得到白色固体2.3g,收率23.8%,uplc纯度98.15%,如图3所示。杂质s的1h核磁谱如图4所示。1h-nmr(600m,dmso-d6)δ=1.40-1.43(m,2h),1.67(quint,4h,j=7.2hz),2.85-2.89(m,2h),3.06-3.22(m,8h),3.28(s,6h),3.36(s,2h),3.50(dd,3h,j=13.2,4.2hz),3.64(s,6h),3.71(s,6h),3.73(s,6h),3.73-3.92(m,8h),4.09-4.14(m,4h),4.85(dd,2h,j=10.2,4.2hz),5.78(s,2h),6.57(dd,2h,j=8.4,2.4hz),6.61(d,2h,j=1.8hz),6.85(m,4h),7.28-7.32(m,6h),7.59-7.61(m,4h)。
[0105]
实施例8
[0106]
式iii化合物(杂质v,1r-反-1's-顺式苯磺酸阿曲库铵)的制备
[0107]
向三口瓶中加入70%苯磺酸水溶液8.0g、纯化水3.5g和式vii化合物(r-反式酯)5.0g(1.0eq),控制釜内温度40
±
3℃搅拌约60分钟,至固体完全溶解,旋蒸蒸除其中水分后,加100ml甲苯和式v化合物5.23g(1.0eq)。装置分水器与回流冷凝管后(分水器中有适量4a分子筛用于吸水),内温控制在25-35℃减压回流反应24小时,静置后分出上层并舍弃,下层用90ml二氯甲烷溶解,用纯化水洗2
×
65ml,经无水硫酸镁干燥后,滴入900ml乙醚中析晶,减压抽滤,得到白色固体6.4g。该粗品经制备液相分离纯化,旋蒸浓缩至约30ml,滴入400ml甲基叔丁基醚中析晶,减压抽滤,将滤饼于35℃下真空干燥6小时,得到白色固体2.0g,收率20.6%,uplc纯度98.31%,如图5所示。核磁图谱如图6所示。1h-nmr(600m,dmso-d6)δ=1.25-1.30(m,2h),1.53(quint,4h,j=7.2hz),2.86-2.90(m,2h),2.98-3.08(m,
8h),3.24(s,6h),3.29(s,6h),3.50-3.59(m,6h),3.64(s,6h),3.67-3.72(m,14h),3.85-3.90(m,2h),3.97(t,4h,j=6.6hz),4.09-4.14(m,4h),4.73(dd,1h,j=10.2,4.2hz),4.93(dd,1h,j=10.2,4.2hz),5.66(s,1h),5.88(s,1h),6.52-6.70(m,4h),6.82-6.84(m,4h),7.28-7.33(m,6h),7.60-7.62(m,4h)。
[0108]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1