番茄根特异性表达启动子pSlNRT2.5及其应用
番茄根特异性表达启动子pslnrt2.5及其应用
技术领域
1.本发明属于生物技术领域,具体涉及番茄根特异性表达启动子pslnrt2.5及其应用。
背景技术:
2.植物基因表达受转录、转录后和翻译水平调控。其中转录水平受启动子调控,启动子在很大程度上决定了基因的表达部位、表达方式、表达时期和表达水平等。启动子包括组成型、组织特异性和诱导型启动子三种类型。近年来,一些强的组成型启动子,例如双子叶植物中表达效率较高的烟草花叶病毒35s(camv 35s)启动子和单子叶植物中常用的玉米ubiquitin 和水稻actin启动子被广泛应用于植物基因工程领域(battraw和hall 1990;mcelroy等1990;christensen等1992)。但组成型启动子表达通常不受外界环境的影响,启动基因持续稳定高效地在所有器官或者组织中表达,不能满足特定基因在特定组织的表达,而且有些目的基因持续高水平在所有组织中表达有可能对寄主植物造成伤害(anami等2013)。随着科学技术的快速发展,特定时期、特定组织以及特定含量地在生物体内表达外源基因,使其更为经济有效的发挥作用,成为基因工程研究的热点。
3.组织特异性启动子,驱动外源基因仅在受体植物的特定组织表达,克服了组成型启动子的缺点。此外,基因在不同发育阶段及组织中表达也是植物本身生长发育的需要。基因的特异表达取决于启动子中存在的顺式作用元件,组织特异性启动子除了具有一般启动子所含有的tata box、caat box和gc box外,还存在控制组织特异性表达所必需的元件,这些元件的种类、数目及相对位置决定了其表达特异性。组织特异性启动子遍布于植物各种组织,包括营养器官特异表达(绿色组织、根)和生殖器官特异表达(雌蕊、花粉、花、种子、胚和胚乳、果实)启动子。在转基因作物育种研究中,利用高效和组织特异性表达启动子是培育高效和安全转基因作物的首选。
4.植物的根是固定植物的根基,是植物吸收水分、无机盐等的唯一通路,也是合成和贮藏营养物质的重要器官,此外根在干旱、盐碱和重金属土壤条件下保护植物地上部分也具有重要作用,因此根在植物整个生命过程中具有举足轻重的作用。根是重要营养器官,了解根特异性表达基因,特别是其启动子对作物改良具有重要的价值。根特异性表达系统主要应用于研究转基因植物在植物抗病虫害、提高植物抵抗土壤致病菌的侵害能力(huang等2006),耐盐碱性、提高植物对恶劣环境的适应能力(gao等2011),以及改变植物代谢途径、提高和改善植物产量、营养成分等方面具有突出的应用价值(xu 等 2010)。已有报道,利用atwrky6根特异表达启动子驱动细胞分裂素氧化酶3(ckx3)在烟草和拟南芥根中特异性表达,根系生物量增加 60%,提高了植株抗干旱和抗重金属胁迫的能力(werner等 2010)。 利用鹰嘴豆wrky31根特异表达基因启动子驱动鹰嘴豆ckx6基因在鹰嘴豆中特异性表达,鹰嘴豆根系更加发达,耐旱能力增强,并增加了种子中矿物质含量(khandal 等 2020)。zhang 等(2016)利用烟草根特异性基因ntrel1启动子驱动大豆白藜芦醇合酶基因(ahrs)在烟草根部特异表达。由此可见,根特异性启动子可以提高植物抗逆性,增加产量,改良品质,具有
良好的发展前景。上述报道利用转基因技术在不同的植物间进行启动子功能分析,表明利用模式植物拟南芥、烟草等作为载体,通过异源转基因分析证实来自其他植物的启动子的功能是被广泛认可和接受的。
[0005] 近年来同时构建几个基因到一个表达载体上的多基因转化系统逐步取代了传统的重复转化和杂交手段用以改良营养品质(lin等2003; wakasa等2006)。在这种多基因表达系统中,每一种基因都需要特异的启动子来驱动表达,以避免由于转入序列同源性过高而引起转基因沉默现象(naqvi等2010)。迄今在番茄中分离的启动子大多是在叶片、种子中特异性表达,有关番茄根特异表达启动子的研究较少,且大多数克隆自已知根特异表达基因,因此分离和鉴定优良的根特异性启动子具有重要作用。发明人前期克隆了sltip、slmt3两个根特异性启动子,但可供选择使用的番茄根特异性启动子还是比较少,尤其是高效表达、片段较小的根特异性启动子。因此,研究番茄根特异性启动子对外源基因表达效率的影响,对在番茄中定向表达外源基因有着重要的理论意义和应用价值。
技术实现要素:
[0006]
本发明的目的是提供一种番茄根特异性启动子pslnrt2.5及其应用,该启动子能够驱动目的基因在植物根中特异性表达,为通过基因工程手段提高外源基因在作物特定组织的表达和积累水平,应用转基因方法获得抗病或抗逆作物新品种奠定了理论基础。
[0007]
为了实现上述目的,本发明采用以下技术方案:番茄根特异性启动子pslnrt2.5,其为具有以下任一的核苷酸序列:1) 序列表中seq id no.1所示的核苷酸序列;2) 与序列表中seq id no.1所示的核苷酸序列有90%以上同源性且具有调控目的基因在植物根中特异性表达功能的核苷酸序列;3) 在高严谨条件下可与序列表中seq id no.1所示的核苷酸序列杂交的核苷酸序列。
[0008]
所述高严谨条件为在0.1
×
sspe (或 0.1
×
ssc),0.1% sds的溶液中,在65℃下杂交并洗膜。
[0009]
扩增上述番茄根特异性启动子pslnrt2.5的引物对,所述引物对的核苷酸序列如seq id no.2和seq id no.3所示。
[0010]
含有上述番茄根特异性启动子pslnrt2.5的基因表达盒。
[0011]
含有上述番茄根特异性启动子pslnrt2.5的表达载体。
[0012]
含有上述番茄根特异性启动子pslnrt2.5的重组菌。
[0013]
上述番茄根特异性启动子pslnrt2.5在启动目的基因表达中的应用。
[0014]
进一步地,所述启动目的基因表达为在植物中启动目的基因表达。
[0015]
更进一步地,所述表达为根特异性表达。
[0016]
利用上述植物表达载体,将本发明的启动子序列构建到任何目的基因上游,导入植物细胞,可获得根特异性表达转基因植株。携带有本发明启动子序列的植物表达载体可通过使用ti质粒、直接dna转化、微注射、农杆菌介导等常规生物学方法转化植物细胞或组织,并将转化的植物组织培育成植株。被转化的植物宿主指拟南芥。
[0017]
与现有技术相比,本发明具有以下优点及有益效果:
1. 该启动子为通过转录组测序技术筛选到的番茄根特异性表达启动子,该方法快速方便、准确率高。
[0018]
2. 该启动子可特异性驱动下游目的基因在植物的根中表达,而不在其他组织、器官中表达,具有组织表达特异性。
[0019]
3. 植物的根是重要营养器官,育种专家可利用slnrt2.5启动子特异性在根中表达目的基因来提高植物抵抗病虫害、耐盐碱性、改变植物代谢途径、提高和改善植物产量、营养成分。slnrt2.5启动子为通过基因工程手段提高外源基因在番茄特定组织的表达和积累水平,应用转基因方法获得抗病或抗逆作物新品种奠定了理论基础。
附图说明
[0020]
图1为番茄slnrt2.5启动子pcr扩增的电泳图谱。
[0021]
图2为番茄slnrt2.5启动子融合gus基因表达载体p1300gn-pslnrt2.5的结构示意图。
[0022]
图3为p1300gn-pslnrt2.5载体双酶切鉴定图谱。
[0023]
图4为转p1300gn-pslnrt2.5载体t1代拟南芥植株pcr扩增电泳图谱。
[0024]
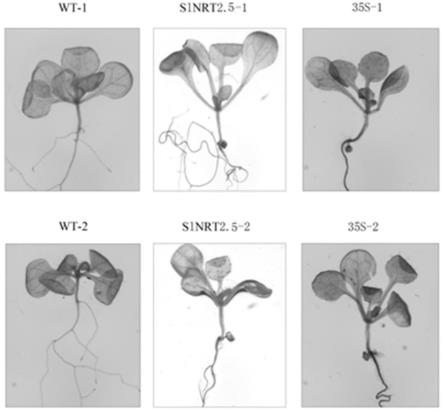
图5为转p1300gn-pslnrt2.5载体t1代拟南芥植株根、地上gus染色结果。
具体实施方式
[0025]
番茄是典型的模式植物,
‘
micro-tom’是一种番茄矮化突变体,既具有番茄的基本特征,也具有植株较矮小,生长周期更短的优点,更适用于功能基因组学研究。本发明根据番茄不同组织部位转录组测序分析筛选番茄根特异表达的启动子,利用 pcr技术克隆番茄根特异性表达基因启动子,并替代pcambia1300gn(p1300gn)中35s启动子与gus报告基因融合,经农杆菌介导的转基因实验获得转基因阳性植株,通过gus组织化学染色和gus酶活检测分析启动子表达部位和启动活性,结合启动子顺式作用元件分析,筛选出活性强烈、作用稳定的番茄根特异性表达启动子pslnrt2.5,为寻找番茄根特异表达启动子提供了有效途径,为通过基因工程手段提高外源基因在作物特定组织的表达和积累水平,应用转基因方法获得抗病或抗逆作物新品种奠定了理论基础。
[0026]
下面结合附图和具体实施例对本发明作进一步详细说明,但不应理解为对本发明的限制。在不背离本发明精神和实质的情况下,对本发明方法、步骤或条件所作的修改或替换,均属于本发明的范围。
[0027]
下述实施例中所用方法如无特别说明均为常规方法,所用引物和测序均由北京美级思诺生物科技有限公司完成,快速内切酶购自thermo fisher scientific,dna凝胶回收试剂盒、dna marker等购自大连宝生物公司;实验中所用的番茄
‘
micro-tom’、野生型拟南芥col-0、大肠杆菌感受态菌株top10、农杆菌感受态菌株gv3101和改造后的植物遗传表达载体p1300gn等,均为发明人实验室保存。
[0028]
实施例1番茄slnrt2.5启动子全长序列的获得通过分析番茄品种heinz花芽、花、根、叶、不同时期果实的官的基因表达转录组数据,筛选到一个在根中特异表达的硝酸盐转运蛋白2家族成员基因slnrt2.5
(solyc11g069735)。根据番茄全基因组序列预测的solyc11g069735序列,选择该基因上游2000bp片段(核苷酸序列如seq id no.1所示)作为该基因的启动子序列,并设计引物,所用上游引物f为acgacggccagtgccaagctttaagaaaatatgacaataaatagc(seq id no.2),下游引物r为ggactgaccacccggggatcctttaggcaaaaaaacaaaaaagtgattaa aatg(seq id no.3)。上游引物f中,前15碱基acgacggccagtgcc为p1300gn载体同源重组臂,其后的aagctt为hindiii酶切位点,其余序列为slnrt2.5上游扩增引物序列;下游引物r中,前15碱基ggactgaccacccgg为p1300gn载体同源重组臂,其后的ggatcc为bamh i酶切位点,其余序列为slnrt2.5下游扩增引物序列。
[0029]
采用 ctab法提取番茄品种
‘
micro-tom’基因组dna,以其作为模板,反应体系如下:5
ꢀµ
l 10
×
缓冲液,2
ꢀµ
l dntps,2
ꢀµ
l dna模板,1
ꢀµ
l dmso,上游引物f和下游引物r各1.5
ꢀµ
l,0.5
ꢀµ
l kod plus-neo聚合酶,ddh2o 36.5
ꢀµ
l。反应条件为:94℃预变性4 min;94℃变性30 s,50~64℃退火30 s,68℃延伸1 min,30个循环;68℃保温10 min。pcr结束后取5μl反应液进行琼脂糖凝胶电泳,结果表明:pcr产物只有1条大小为2000bp的dna带。图1中第1泳道为dl5000 marker,条带从上至下分别为5000 bp、3000 bp、2000bp、1000 bp、750 bp、500 bp、250bp、100 bp。第2泳道为slnrt2.5启动子,将此片段回收纯化。
[0030]
实验例2slnrt2.5启动子植物表达载体构建使用thermo fisher scientific hind
ꢀⅲ
和bamh i双酶切p1300gn,双酶切体系为:hind
ꢀⅲꢀ2ꢀµ
l,bamh i 2
ꢀµ
l,p1300gn质粒20
ꢀµ
l,fastdigest绿色缓冲液 4
ꢀµ
l,ddh2o 12
ꢀµ
l。37℃酶切1 h,酶切产物琼脂糖凝胶电泳后参照dna凝胶回收试剂盒说明书进行纯化。将纯化的pcr片段与酶切回收的空载体通过同源重组法连接,构建含有gus基因的植物表达载体p1300gn-pslnrt2.5(图2)。连接体系为:载体骨架2.5
µ
l,纯化pcr片段2.5
µ
l,2
×
easygeno assembly mix 5
µ
l,50℃水浴30min。之后将10
µ
l连接产物加入到含有50
µ
l感受态细胞top10的离心管中,温和混匀,冰浴30min,42℃热激90s,冰浴2min,加入800
µ
l lb液体培养基,于37℃培养45min,取200
µ
l菌液涂布于含50
µ
g/l卡那霉素的lb培养基平皿中,37℃倒置培养12~16h,观察结果。菌落pcr 反应体系:1 μl引物 f,1 μl引物 r,10 μl 2
×
a8 mixture,单菌落,ddh2o 8 μl。pcr反应条件为:95℃预变性5 min;95℃变性30 s,56℃退火30 s,72℃延伸2 min,循环35次;72℃保温5 min。鉴定呈阳性的克隆提取质粒dna,进行双酶切检测,酶切正确载体送往北京美级思诺生物科技有限公司测序。双酶切验证及测序结果正确的重组子命名为p1300gn
‑ꢀ
pslnrt2.5(图3)。图3中第1泳道为dl5000 marker,第2泳道为重组子p1300gn
‑ꢀ
pslnrt2.5,第3泳道为p1300gn
‑ꢀ
pslnrt2.5hind
ꢀⅲ
和bamh i双酶切结果。
[0031]
实验例3拟南芥的遗传转化及阳性植株检测采用热激法将构建好的双元表达载体p1300gn-pslnrt2.5及p1300gn对照载体(含35s启动子)导入农杆菌gv3101。将待转化的拟南芥植株去顶,剪去已经开放的花蕾,进行转化。取含有p1300gn-pslnrt2.5质粒和p1300gn质粒的农杆菌gv3101菌液20 μl加到3 ml lb(50 μg/ml kan)液体培养基中,28℃,180 r/min培养36-48 h;菌液置于离心管中,室温12000 r/min离心1 min收集菌体,弃上清;加1 ml 1/2ms+5%蔗糖溶液悬浮沉淀,12000 r/
min离心1 min,弃上清;再次加1 ml 1/2ms+5%蔗糖溶液悬浮沉淀后加0.2 μl 0.02% silwet-l77,弹拨混匀;用200 μl移液器吸取菌体至未开放花蕾上,并做好标记,用塑料膜覆盖过夜,提高转化率。大约一个月后种子成熟,收获后烘干,置于4℃春化2d,标注为t0代种子。使用10%naclo消毒10 mint0代拟南芥种子,灭菌水清洗5次,点种于含有40μg/ml潮霉素的ms培养基表面,4℃冰箱春化2天,再置于23℃培养间培养。
[0032]
提取野生型、转p1300gn-pslnrt2.5质粒和p1300gn拟南芥基因组 dna,并选用gus为目标基因进行扩增。以植物表达载体引物gus f:atgttacgtcctgtagaaacc(seq id no.4)和gus r:cggcaataacatacggcgtgacatc(seq id no.5)来筛选阳性转基因植株,pcr 扩增反应体系为:1 μl 模板,0.5 μl gus f,0.5 μl gus r,10 μl 2
×
a8 mixture, ddh2o 8 μl。经pcr检测为阳性的转基因材料为t1代(如图4所示)。图4中第1泳道(标m)为dl2000 marker,分子量大小分别为2000 bp、1000 bp、750 bp、500 bp、250 bp、100 bp。第2泳道(标1)为以质粒为模板的阳性对照,第3泳道(标2)为以野生型植株为模板的阴性对照,第4~15泳道(标3-14)为不同的p1300-pslnrt2.5转基因拟南芥植株。t0代转基因植株所结的种子和由该种子长成的植株为t1代,依此类推,t2、t3分别为转基因植株第2代和第3代。
[0033]
实验例4转基因植株的gus组织化学染色采用组织化学染色法检测gus基因在植株组织细胞中的表达。取实施例3中获得的t1代转基因拟南芥幼苗进行gus组织化学染色,分别以野生型和转p1300gn(含35s启动子)对照载体拟南芥作为阴性和阳性对照。将待测拟南芥植株用无菌水冲洗干净,吸取表面水分,分别放于2 ml离心管中,每管加入适量的gus染色液(gus染色试剂盒sl7160-2购自coolaber)于37℃温育1 h及以上,除去染液,用70%乙醇脱色至阴性对照材料叶片呈白色,使用stereo discovery v.12扫描拍照。白色背景下的蓝色即为gus表达位点。结果显示,野生型拟南芥的根、地上部分未见蓝色,含35s启动子对照载体转化植株的根及地上部分均呈蓝色,而转化重组载体p1300gn-pslroot1植株地上部分始终检测不到gus表达,仅在根中有强烈特异表达(如图5所示)。因此该启动子驱动的gus基因仅在拟南芥根中高效表达,在其他组织和器官中均不表达,即该启动子为根特异性启动子,在植物基因工程中具有良好的应用价值。如可以通过构建含有该启动子驱动提高植株抗逆、抗倒伏的载体,转化受体植株,人工创造抗性强、优质高产材料,应用于农业生产。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1