一种N-杂环BET溴结构域抑制剂的合成方法及其中间体与流程
一种n-杂环bet溴结构域抑制剂的合成方法及其中间体
技术领域
1.本发明属于药物化学合成技术领域,具体涉及化合物iii、化合物iv和化合物vi及其在制备一种n-杂环bet溴结构域抑制剂(化合物vii)中的用途。
背景技术:
2.组蛋白乙酰化是表观遗传研究的重要组成部分,乙酰化的组蛋白可通过dna聚合酶与rna聚合酶及转录因子作用激活基因转录。溴结构域和超末端结构域(bet)家族属于溴结构域蛋白家族(bromodomain proteins,brds),是一类进化上高度保守的蛋白,其可识别并结合组蛋白尾部的乙酰化赖氨酸残基,招募染色质调节相关蛋白、转录因子、染色质重塑因子等,从而在调控基因转录和染色质重塑中发挥重要作用,其与细胞生长、增殖分化、凋亡坏死等多种生物学过程相关,是重要的表观遗传“阅读器”。
3.目前为止,已发现人体基因组共编码61种溴结构域,分布在46种不同的蛋白质中。brds家族由brd2、brd3、brd4、brdt 4个亚型组成,它们均含有两个串联的溴结构域(bd1、bd2)和一个超末端(et)结构域。两个溴结构域主要负责识别并结合乙酰化赖氨酸残基,et结构域则与辅助因子相互作用。bet蛋白的异常表达与多种疾病关联,虽然bet家族的4个成员具有相似的结构,但其生物学功能仍存在差异,尤其是brd4与癌症和炎症等多种疾病密切相关(jung m等,epigenomics,2015,7(3):487-501.)。
4.靶向bet蛋白对于发展靶向癌症、炎症和病毒的新的治疗策略是有益的。目前已有针对bet蛋白的小分子抑制剂进入临床前和临床研究阶段,其主要用于癌症及自身免疫疾病的治疗。
5.综上,bet蛋白抑制剂作为药物研发具有很好的应用前景,申请人成都苑东生物制药有限公司开发了一种表观遗传学相关的溴结合域蛋白brd4抑制剂,化学名称为:n-乙基-4-(5-(2-羟基丙基-2-基)-3-(吲哚啉-1-基)噻吩-2-基)-6-甲基-7-氧代-6,7-二氢-1h-吡咯并[2,3-c]吡啶-2-甲酰胺,其结构式如式vii所示,申请人在先申请的专利pct/cn2021/134454(申请日2021.11.30,国内申请号202180005165.6)中报道了该化合物具有较好的bet蛋白抑制活性及良好的药代动力学性质,同时可表现出良好的体内抑瘤活性且herg心脏毒性风险低,为新一代高效低毒的bet蛋白抑制剂。
[0006][0007]
因此,针对式vii化合物开发一条工艺简单、生产成本低、环境污染小,适合工业化生产的制备方法具有重要现实意义。
技术实现要素:
[0008]
本发明的第一个目的在于提供一种化合物iii,具有如下结构:
[0009][0010]
其中,r选自c1-c6烷基;
[0011]
优选地,r选自c1-c4烷基;
[0012]
进一步优选地,r选自甲基或乙基;
[0013]
更进一步优选地,r选自甲基。
[0014]
本发明的第二个目的在于提供一种化合物iv,具有如下结构:
[0015][0016]
其中,r选自c1-c6烷基;
[0017]
优选地,r选自c1-c4烷基;
[0018]
进一步优选地,r选自甲基或乙基;
[0019]
更进一步优选地,r选自甲基。
[0020]
本发明的第三个目的在于提供一种化合物vi,具有如下结构:
[0021][0022]
其中,r选自c1-c6烷基;
[0023]
优选地,r选自c1-c4烷基;
[0024]
进一步优选地,r选自甲基或乙基;
[0025]
更进一步优选地,r选自甲基。
[0026]
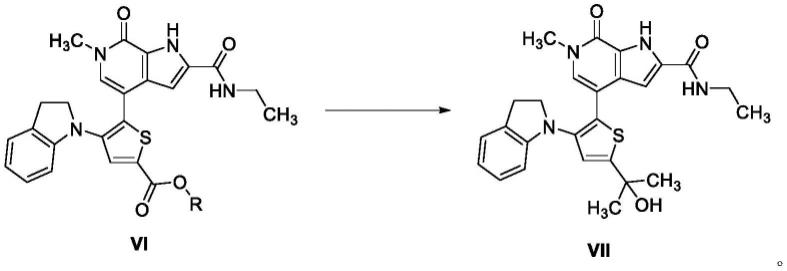
本发明的第四个目的在于提供一种制备化合物vii的方法,该方法包括如下步骤:化合物vi与甲基金属试剂在溶剂d中反应制备化合物vii
[0027][0028]
其中,甲基金属试剂选自甲基锂、甲基溴化镁、甲基氯化镁、甲基碘化镁、甲基锌或甲基铝,优选为甲基锂或甲基溴化镁;
[0029]
其中,化合物vi与甲基金属试剂的摩尔比为1:4.0~4.5;
[0030]
其中,溶剂d选自四氢呋喃、2-甲基四氢呋喃,优选四氢呋喃;
[0031]
其中,反应温度为0~30℃,优选10~20℃。
[0032]
在一些具体的实施方案中,本发明制备化合物vii的方法,包括如下步骤:
[0033]
(1)化合物iv与化合物v在催化剂b、碱2和溶剂c的作用下反应制备化合物vi
[0034][0035]
在一些具体的实施方案中,所述反应中还包括催化剂b的配体,其中,催化剂b的配体选自x-phos、s-phos、asph3、n-bu3p、(meo)3p、pph3、以及双齿配体ph2p(ch2)2pph2(dppe)或ph2p(ch2)3pph2(dppp),优选为pph3、asph3或双齿配体ph2p(ch2)2pph2(dppe)、ph2p(ch2)3pph2(dppp),更优选为pph3;
[0036]
其中,催化剂b与催化剂b配体的摩尔比为1:1.0-4.0,优选为1:1.0-1.5;
[0037]
其中,化合物iv与化合物v的摩尔比为1.0:0.9~1.2,优选1.0:1.0;
[0038]
其中,催化剂b选自钯催化剂四(三苯基膦)钯、醋酸钯、二(三苯基膦)二氯化钯或1,1-双(二苯基膦)二荗铁二氯化镍,优选为四(三苯基膦)钯、醋酸钯,更优选为四(三苯基膦)钯;
[0039]
其中,化合物iv与催化剂b的摩尔比为1:0.05-0.3,优选为1:0.05-0.10;
[0040]
其中,碱2选自碳酸钾,碳酸钠,碳酸铯,磷酸钾,优选碳酸钾;
[0041]
其中,化合物iv与碱2的摩尔比为1:2.0-3.5;
[0042]
其中,反应溶剂c为有机溶剂和水组成的混合溶剂,所述有机溶剂选自1,4-二氧六环、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲亚砜、n-甲基吡咯烷酮,优选为1,4-二氧六环、n,n-二甲基乙酰胺,更优选为1,4-二氧六环;
[0043]
其中,混合溶剂中有机溶剂与水的质量比为1:0.05~0.20;
[0044]
其中,反应温度为60~120℃,优选为80~120℃,更优选为80~90℃;
[0045]
(2)化合物vi与甲基金属试剂在溶剂d中反应制备化合物vii
[0046][0047]
其中,甲基金属试剂选自甲基锂、甲基溴化镁、甲基氯化镁、甲基碘化镁、甲基锌或甲基铝,优选为甲基锂或甲基溴化镁;
[0048]
其中,化合物vi与甲基金属试剂的摩尔比为1:4.0~4.5;
[0049]
其中,溶剂d选自四氢呋喃、2-甲基四氢呋喃,优选四氢呋喃;
[0050]
其中,反应温度为0~30℃,优选10~20℃。
[0051]
在一些具体的实施方案中,本发明制备化合物vii的方法,包括如下步骤:
[0052]
(1)化合物iii与溴化试剂在溶剂b中反应制备化合物iv
[0053][0054]
其中,溴化试剂选自溴素、nbs、三溴吡啶或二溴海因,优选nbs或二溴海因,更优选为nbs;
[0055]
其中,化合物iii与溴化试剂的摩尔比为1.0:0.9-1.2,优选1.0:1.0;
[0056]
其中,溶剂b为有机溶剂,所述有机溶剂选自乙腈、乙酸乙酯、丙酮、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺或n-甲基吡咯烷酮,优选乙腈、丙酮,更优选为乙腈;
[0057]
其中,反应温度为-10~30℃,优选0~20℃,更优选为0~10℃;
[0058]
(2)化合物iv与化合物v在催化剂b、碱2和溶剂c的作用下反应制备化合物vi
[0059][0060]
在一些具体的实施方案中,所述反应中还包括催化剂b的配体,其中,催化剂b的配体选自x-phos、s-phos、asph3、n-bu3p、(meo)3p、pph3、以及双齿配体ph2p(ch2)2pph2(dppe)或ph2p(ch2)3pph2(dppp),优选为pph3、asph3或双齿配体ph2p(ch2)2pph2(dppe)、ph2p(ch2)3pph2(dppp),更优选为pph3;
[0061]
其中,催化剂b与催化剂b配体的摩尔比为1:1.0-4.0,优选为1:1.0-1.5;
[0062]
其中,化合物iv与化合物v的摩尔比为1.0:0.9~1.2,优选1.0:1.0;
[0063]
其中,催化剂b选自钯催化剂四(三苯基膦)钯、醋酸钯、二(三苯基膦)二氯化钯或1,1-双(二苯基膦)二荗铁二氯化镍,优选为四(三苯基膦)钯、醋酸钯,更优选为四(三苯基膦)钯;
[0064]
其中,化合物iv与催化剂b的摩尔比为1:0.05-0.3,优选为1:0.05-0.10;
[0065]
其中,碱2选自碳酸钾,碳酸钠,碳酸铯,磷酸钾,优选碳酸钾;
[0066]
其中,化合物iv与碱2的摩尔比为1:2.0-3.5;
[0067]
其中,反应溶剂c为有机溶剂和水组成的混合溶剂,所述有机溶剂选自1,4-二氧六环、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲亚砜、n-甲基吡咯烷酮,优选为1,4-二氧六环、n,n-二甲基乙酰胺,更优选为1,4-二氧六环;
[0068]
其中,混合溶剂中有机溶剂与水的质量比为1:0.05~0.20;
[0069]
其中,反应温度为60~120℃,优选为80~120℃,更优选为80~90℃;
[0070]
(3)化合物vi与甲基金属试剂在溶剂d中反应制备化合物vii
[0071][0072]
其中,甲基金属试剂选自甲基锂、甲基溴化镁、甲基氯化镁、甲基碘化镁、甲基锌或甲基铝,优选为甲基锂或甲基溴化镁;
[0073]
其中,化合物vi与甲基金属试剂的摩尔比为1:4.0~4.5;
[0074]
其中,溶剂d选自四氢呋喃、2-甲基四氢呋喃,优选四氢呋喃;
[0075]
其中,反应温度为0~30℃,优选10~20℃。
[0076]
在一些具体的实施方案中,本发明制备化合物vii的方法,包括如下步骤:
[0077]
(1)化合物i与化合物ii在催化剂a、碱1和溶剂a的作用下反应制备化合物iii
[0078][0079]
x为cl、br或i;
[0080]
在一些具体的实施方案中,所述反应中还包括催化剂a的配体;
[0081]
其中,催化剂a的配体选自p(o-tolyl)3、p(t-bu)3、cypf-t-bu、josiphos、binap、xantphos、dppf、brett-phos、ru-phos、x-phos、s-phos、bippy-phos,优选为josiphos、xantphos、brett-phos、ru-phos、x-phos、s-phos、bippy-phos或binap;更优选为s-phos、binap;
[0082]
其中,化合物i与化合物ii的摩尔比为1:1.0~2.0,优选1:1.2;
[0083]
其中,催化剂a选自四(三苯基膦)钯、三(双亚苄基丙酮)二钯、醋酸钯和1,1-双(二
苯基膦)二荗铁二氯化钯中的一种,优选为三(双亚苄基丙酮)二钯、醋酸钯,更优选为三(双亚苄基丙酮)二钯;
[0084]
其中,化合物i与催化剂a的摩尔比为1:0.025~0.05;
[0085]
其中,催化剂a与催化剂a配体的摩尔比为1:1.0~1.5;
[0086]
其中,碱1选自叔丁醇钾、叔丁醇钠、碳酸钾、碳酸钠、碳酸铯、碳酸锂、磷酸钾、氢氧化钾、氢氧化钠,优选为叔丁醇钾、叔丁醇钠、碳酸钾、碳酸钠、碳酸铯、碳酸锂、磷酸钾,更优选为叔丁醇钠、碳酸铯;
[0087]
其中,化合物i与碱1的摩尔比为1:2.0~3.5,优选1:2.0;
[0088]
其中,反应溶剂a为有机溶剂,所述有机溶剂选自甲苯、二甲苯、四氢呋喃、2-甲基四氢呋喃或1,4-二氧六环,优选为甲苯、二甲苯、四氢呋喃或2-甲基四氢呋喃,更优选为甲苯或四氢呋喃;
[0089]
其中,反应温度为60~120℃,优选60~80℃,更优选为60~75℃;
[0090]
(2)化合物iii与溴化试剂在溶剂b中反应制备化合物iv
[0091][0092]
其中,溴化试剂选自溴素、nbs、三溴吡啶或二溴海因,优选nbs或二溴海因,更优选为nbs;
[0093]
其中,化合物iii与溴化试剂的摩尔比为1.0:0.9-1.2,优选1.0:1.0;
[0094]
其中,溶剂b为有机溶剂,所述有机溶剂选自乙腈、乙酸乙酯、丙酮、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺或n-甲基吡咯烷酮,优选乙腈、丙酮,更优选为乙腈;
[0095]
其中,反应温度为-10~30℃,优选0~20℃,更优选为0~10℃;
[0096]
(3)化合物iv与化合物v在催化剂b、碱2和溶剂c的作用下反应制备化合物vi
[0097][0098]
在一些具体的实施方案中,所述反应中还包括催化剂b的配体,其中,催化剂b的配体选自x-phos、s-phos、asph3、n-bu3p、(meo)3p、pph3、以及双齿配体ph2p(ch2)2pph2(dppe)或ph2p(ch2)3pph2(dppp),优选为pph3、asph3或双齿配体ph2p(ch2)2pph2(dppe)、ph2p(ch2)3pph2(dppp),更优选为pph3;
[0099]
其中,催化剂b与催化剂b配体的摩尔比为1:1.0-4.0,优选为1:1.0-1.5;
[0100]
其中,化合物iv与化合物v的摩尔比为1.0:0.9~1.2,优选1.0:1.0;
[0101]
其中,催化剂b选自钯催化剂四(三苯基膦)钯、醋酸钯、二(三苯基膦)二氯化钯或1,1-双(二苯基膦)二荗铁二氯化镍,优选为四(三苯基膦)钯、醋酸钯,更优选为四(三苯基
膦)钯;
[0102]
其中,化合物iv与催化剂b的摩尔比为1:0.05-0.3,优选为1:0.05-0.10;
[0103]
其中,碱2选自碳酸钾,碳酸钠,碳酸铯,磷酸钾,优选碳酸钾;
[0104]
其中,化合物iv与碱2的摩尔比为1:2.0-3.5;
[0105]
其中,反应溶剂c为有机溶剂和水组成的混合溶剂,所述有机溶剂选自1,4-二氧六环、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲亚砜、n-甲基吡咯烷酮,优选为1,4-二氧六环、n,n-二甲基乙酰胺,更优选为1,4-二氧六环;
[0106]
其中,混合溶剂中有机溶剂与水的质量比为1:0.05~0.20;
[0107]
其中,反应温度为60~120℃,优选为80~120℃,更优选为80~90℃;
[0108]
(4)化合物vi与甲基金属试剂在溶剂d中反应制备化合物vii
[0109][0110]
其中,甲基金属试剂选自甲基锂、甲基溴化镁、甲基氯化镁、甲基碘化镁、甲基锌或甲基铝,优选为甲基锂或甲基溴化镁;
[0111]
其中,化合物vi与甲基金属试剂的摩尔比为1:4.0~4.5;
[0112]
其中,溶剂d选自四氢呋喃、2-甲基四氢呋喃,优选四氢呋喃;
[0113]
其中,反应温度为0~30℃,优选10~20℃。
[0114]
在一些具体的实施方案中,本发明制备化合物vii的方法,包括如下步骤:
[0115][0116]
(1)化合物i、化合物ii以及碳酸铯、三(双亚苄基丙酮)二钯、s-phos在甲苯中,于75℃下反应生成化合物iii;
[0117]
(2)化合物iii与nbs于0~10℃下,在乙腈中反应生成化合物iv;
[0118]
(3)化合物iv与化合物v发生suzuki反应得到化合物vi;
[0119]
(4)化合物vi与甲基锂在四氢呋喃中,于10~20℃下反应生成化合物vii。
[0120]
本发明取得的有益效果:
[0121]
本发明方法采用化合物i(4-溴噻吩-2-甲酸甲酯)、化合物ii(吲哚啉)和化合物v(2-(乙氨基羰基)-6-甲基-7-氧代-6,7-二氢-1h-吡咯并[2,3-c]吡啶-4-硼酸频那醇酯)作为原料,经过4步反应即得到目标化合物vii;本发明起始原料简单易购,成本较低,每步反应条件温和,操作简单,收率高,无需使用柱层析和冻干技术;同时,通过本发明的制备方法所得产品纯度大于99%,且总收率高,因此,有利于实现工业化生产。
具体实施方式
[0122]
以下结合实施例对本发明作进一步的详细描述,但并非对本发明的限制,凡依照本发明公开内容所作的任何本领域的等同替换,均属于本发明的保护范围。
[0123]
化合物的结构是通过质谱(ms)或核磁共振(1hnmr、
13
c nmr)来确定的。
[0124]
核磁共振(1hnmr、
13
c nmr)位移(δ)以百万分之一(ppm)的单位给出;核磁共振(1hnmr)的测定是用brukeravance-400核磁仪,测定溶剂为氘代二甲亚砜(dmso),内标为四甲基硅烷(tms),化学位移是以10-6
(ppm)作为单位给出。
[0125]
质谱(ms)的测定用finnigan lcqad(esi)质谱仪(生产商:therm,型号:finnigan lcq advantage max)进行。
[0126]
在本发明未给出特殊说明的情况下,本发明反应中提及的溶液是水溶液。
[0127]
在本发明的术语“室温”是指温度处于10℃~25℃之间。
[0128]
在本发明中,除非另有说明,使用的术语“cm-cn”是指由该术语修饰的该部分中具有m-n个碳原子(n大于m,且二者为整数)。例如,c1-c6表示其修饰的部分中具有1-6个碳原子,例如1个碳原子、2个碳原子、3个碳原子、4个碳原子、5个碳原子或6个碳原子。
[0129]
在发明中,除非另有说明,使用的术语“烷基”是指仅由碳原子和氢原子组成的饱和烃基,包括但不限于,c1-c6烷基、c1-c5烷基、c1-c4烷基、c1-c3烷基、c1-c2烷基和c1烷基。作为烷基的非限制性实例,可以列举以下直链或支链的饱和烃基:甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基及其另外七种异构体、正己基及其另外十六种异构体。例如,c1-c7烷基包括甲基、乙基、丙基、丁基、戊基、己基、庚基及其全部异构体。
[0130]
词语“包括”、“包含”和“含有”及其等同物应理解为开放的、非排他性的意义,即“包括但不限于”,意味着除所列出的要素、组分和步骤外,还可涵盖其它未指明的要素、组分和步骤。
[0131]
实施例1化合物iii 4-(吲哚啉-1-基)噻吩-2-羧酸甲酯的制备
[0132][0133]
在室温下,将甲苯1l,化合物i 100g,化合物ii 64.5g,碳酸铯221.15g加入反应瓶,氮气置换,再加入s-phos 9.25g和三(双亚苄基丙酮)二钯10.35g,再次氮气置换,升温
至75℃反应20小时。降温至室温,加入水1l,抽滤,分液。有机相用1.2l 1m盐酸洗涤两次,再用饱和盐水洗涤,分液,减压浓缩掉有机溶剂。向残留物中加入甲醇400ml,室温搅拌2小时,抽滤,得到化合物iii的粗品。用乙腈重结晶纯化,干燥,得纯的化合物iii 74.8g(hplc纯度99.50%,收率64%)。
[0134]
ms
+
[m+1]260.2。
[0135]1h nmr(400mhz,dmso-d6)δ7.77(d,j=1.9hz,1h),7.31(d,j=1.9hz,1h),7.16(dd,j=7.2,1.4hz,1h),7.09(td,j=7.6,1.4hz,1h),7.05
–
6.97(m,1h),6.74(td,j=7.3,1.1hz,1h),3.92(t,j=8.5hz,2h),3.84(s,3h),3.09(t,j=8.5hz,2h).
[0136]
13
c nmr(101mhz,dmso-d6)δ161.80,146.10,142.88,131.42,130.51,127.29,125.26,124.95,119.05,110.93,110.89,107.73,52.30,51.98,27.35.
[0137]
实施例2化合物iii 4-(吲哚啉-1-基)噻吩-2-羧酸甲酯的制备
[0138]
在室温下,将四氢呋喃100ml,化合物i10g,化合物ii 6.45g,碳酸铯22.12g加入反应瓶,氮气置换,再加入s-phos 0.93g和三(双亚苄基丙酮)二钯1.04g,再次氮气置换,升温至75℃反应20小时。降温至室温,加入乙酸乙酯100ml和水100ml,抽滤,分液。有机相用120ml 1m盐酸洗涤两次,再用饱和盐水洗涤,分液,减压浓缩掉有机溶剂。向残留物中加入甲醇40ml,室温搅拌2小时,抽滤,得到化合物iii的粗品。用乙腈重结晶纯化,干燥,得纯的化合物iii 6.25g(hplc纯度99.17%,收率53.3%)。
[0139]
实施例3化合物iii 4-(吲哚啉-1-基)噻吩-2-羧酸甲酯的制备
[0140]
在室温下,将甲苯100ml,化合物i10g,化合物ii 10.78g,碳酸铯22.12g加入反应瓶,氮气置换,再加入s-phos 0.93g和三(双亚苄基丙酮)二钯1.04g,再次氮气置换,升温至75℃反应20小时。降温至室温,加入水100ml,抽滤,分液。有机相用120ml 1m盐酸洗涤两次,再用饱和盐水洗涤,分液,减压浓缩掉有机溶剂。向残留物中加入甲醇40ml,室温搅拌2小时,抽滤,得到化合物iii的粗品。用乙腈重结晶纯化,干燥,得纯的化合物iii 7.54g(hplc纯度99.68%,收率64.3%)。
[0141]
实施例4化合物iii 4-(吲哚啉-1-基)噻吩-2-羧酸甲酯的制备
[0142]
在室温下,将四氢呋喃100ml,化合物i10g,化合物ii 6.45g,碳酸铯22.12g加入反应瓶,氮气置换,再加入binap1.42g和三(双亚苄基丙酮)二钯1.04g,再次氮气置换,升温至75℃反应20小时。降温至室温,加入乙酸乙酯100ml和水100ml,抽滤,分液。有机相用120ml1m盐酸洗涤两次,再用饱和盐水洗涤,分液,减压浓缩掉有机溶剂。向残留物中加入甲醇40ml,室温搅拌2小时,抽滤,得到化合物iii的粗品。用乙腈重结晶纯化,干燥,得纯的化合物iii 4.32g(hplc纯度99.36%,收率36.9%)。
[0143]
实施例5化合物iii 4-(吲哚啉-1-基)噻吩-2-羧酸甲酯的制备
[0144]
在室温下,将甲苯100ml,化合物i10g,化合物ii 10.78g,叔丁醇钠6.5g加入反应瓶,氮气置换,再加入s-phos 0.93g和三(双亚苄基丙酮)二钯1.04g,再次氮气置换,升温至75℃反应20小时。降温至室温,加入水100ml,抽滤,分液。有机相用120ml 1m盐酸洗涤两次,再用饱和盐水洗涤,分液,减压浓缩掉有机溶剂。柱层析纯化,得纯的化合物iii 4.17g(hplc纯度98.57%,收率35.6%)。
[0145]
实施例6化合物iii 4-(吲哚啉-1-基)噻吩-2-羧酸甲酯的制备
[0146]
在室温下,将甲苯100ml,化合物i10g,化合物ii 6.45g,碳酸铯22.12g加入反应
瓶,氮气置换,再加入s-phos 1.85g和三(双亚苄基丙酮)二钯2.07g,再次氮气置换,升温至75℃反应14小时。降温至室温,加入水100ml,抽滤,分液。有机相用120ml 1m盐酸洗涤两次,再用饱和盐水洗涤,分液,减压浓缩掉有机溶剂。向残留物中加入甲醇40ml,室温搅拌2小时,抽滤,得到化合物iii的粗品。用乙腈重结晶纯化,干燥,得纯的化合物iii 7.31g(hplc纯度99.08%,收率62.3%)。
[0147]
实施例7化合物iii 4-(吲哚啉-1-基)噻吩-2-羧酸甲酯的制备
[0148]
在室温下,将甲苯100ml,化合物i10g,化合物ii 6.45g,碳酸铯38.70g加入反应瓶,氮气置换,再加入s-phos 0.93g和三(双亚苄基丙酮)二钯1.04g,再次氮气置换,升温至75℃反应20小时。降温至室温,加入水100ml,抽滤,分液。有机相用120ml 1m盐酸洗涤两次,再用饱和盐水洗涤,分液,减压浓缩掉有机溶剂。向残留物中加入甲醇40ml,室温搅拌2小时,抽滤,得到化合物iii的粗品。用乙腈重结晶纯化,干燥,得纯的化合物iii 7.68g(hplc纯度99.34%,收率65.4%)。
[0149]
实施例8化合物iii 4-(吲哚啉-1-基)噻吩-2-羧酸甲酯的制备
[0150]
在室温下,将甲苯100ml,化合物i10g,化合物ii 6.45g,碳酸铯22.12g加入反应瓶,氮气置换,再加入s-phos 0.93g和三(双亚苄基丙酮)二钯1.04g,再次氮气置换,升温至60℃反应41小时。降温至室温,加入水100ml,抽滤,分液。有机相用120ml 1m盐酸洗涤两次,再用饱和盐水洗涤,分液,减压浓缩掉有机溶剂。向残留物中加入甲醇40ml,室温搅拌2小时,抽滤,得到化合物iii的粗品。用乙腈重结晶纯化,干燥,得纯的化合物iii 5.83g(hplc纯度99.42%,收率49.7%)。
[0151]
实施例9化合物iii 4-(吲哚啉-1-基)噻吩-2-羧酸甲酯的制备
[0152]
在室温下,将甲苯100ml,化合物i10g,化合物ii 6.45g,碳酸铯22.12g加入反应瓶,氮气置换,再加入s-phos 0.93g和三(双亚苄基丙酮)二钯1.04g,再次氮气置换,升温至120℃反应10小时。降温至室温,加入水100ml,抽滤,分液。有机相用120ml 1m盐酸洗涤两次,再用饱和盐水洗涤,分液,减压浓缩掉有机溶剂。向残留物中加入甲醇40ml,室温搅拌2小时,抽滤,得到化合物iii的粗品。用乙腈重结晶纯化,干燥,得纯的化合物iii 6.17g(hplc纯度98.93%,收率52.6%)。
[0153]
实施例10化合物iv 4-(吲哚啉-1-基)-5-溴噻吩-2-羧酸甲酯的制备
[0154][0155]
在室温下,将乙腈675ml、化合物iii 45g加入反应瓶中,开启搅拌,降温至5℃,再加入29.2g nbs溶解于450ml乙腈的溶液,于5℃反应1小时。抽滤,30℃降压干燥4h,再用乙腈重结晶,得纯的化合物iv 45.6g(hplc纯度99.21%,收率77.6%)。
[0156]
ms
+
[m+1]338.0/340.0。
[0157]1h nmr(400mhz,dmso-d6)δ7.76(s,1h),7.17(dd,j=7.2,1.3hz,1h),7.01(td,j=7.7,1.3hz,1h),6.73(t,j=7.5hz,1h),6.37(d,j=7.8hz,1h),3.87(t,j=8.4hz,2h),3.82(s,3h),3.10(t,j=8.4hz,2h).
[0158]
13
c nmr(101mhz,dmso-d6)δ160.65,148.12,143.60,131.93,129.63,129.13,126.98,124.84,119.15,111.76,108.51,53.16,52.58,28.18.
[0159]
实施例11化合物iv 4-(吲哚啉-1-基)-5-溴噻吩-2-羧酸甲酯的制备
[0160]
在室温下,将乙腈675ml、化合物iii 45g加入反应瓶中,开启搅拌,降温至5℃,再加入49.6g二溴海因溶解于450ml乙腈的溶液,于5℃反应1小时。抽滤,30℃降压干燥4h,再用乙腈重结晶,得纯的化合物iv 32.6g(hplc纯度98.43%,收率55.5%)。
[0161]
实施例12化合物iv 4-(吲哚啉-1-基)-5-溴噻吩-2-羧酸甲酯的制备
[0162]
在室温下,将乙腈675ml、化合物iii 45g加入反应瓶中,开启搅拌,降温至5℃,再加入35.04g nbs溶解于450ml乙腈的溶液,于5℃反应1小时。抽滤,30℃降压干燥4h,再用乙腈重结晶,得纯的化合物iv 40.08g(hplc纯度98.67%,收率68.3%)。
[0163]
实施例13化合物iv 4-(吲哚啉-1-基)-5-溴噻吩-2-羧酸甲酯的制备
[0164]
在室温下,将丙酮675ml、化合物iii 45g加入反应瓶中,开启搅拌,降温至5℃,再加入29.2g nbs溶解于450ml丙酮的溶液,于5℃反应1小时。加水100ml,搅拌30min,抽滤,30℃降压干燥4h,再用乙腈重结晶,得纯的化合物iv 43.9g(hplc纯度99.17%,收率74.8%)。
[0165]
实施例14化合物iv 4-(吲哚啉-1-基)-5-溴噻吩-2-羧酸甲酯的制备
[0166]
在室温下,将乙腈675ml、化合物iii 45g加入反应瓶中,开启搅拌,降温至0℃,再加入29.2g nbs溶解于450ml乙腈的溶液,于0℃反应1小时。抽滤,30℃降压干燥4h,再用乙腈重结晶,得纯的化合物iv 44.6g(hplc纯度99.59%,收率76.0%)。
[0167]
实施例15化合物iv 4-(吲哚啉-1-基)-5-溴噻吩-2-羧酸甲酯的制备
[0168]
在室温下,将乙腈675ml、化合物iii 45g加入反应瓶中,开启搅拌,降温至10℃,再加入29.2g nbs溶解于450ml乙腈的溶液,于10℃反应1小时。抽滤,30℃降压干燥4h,再用乙腈重结晶,得纯的化合物iv 46.1g(hplc纯度98.86%,收率78.5%)。
[0169]
实施例16化合物vi n-乙基-4-[5-甲氧基羰基-3-(吲哚啉-1-基)噻吩-2-基]-6-甲基-7-氧代-6,7-二氢-1h-吡咯并[2,3-c]吡啶-2-甲酰胺的制备
[0170][0171]
在室温下,将1,4-二氧六环405ml、水45ml、化合物iv 15.0g、化合物v18.3g、碳酸钾18.3g和四(三苯基膦)钯3.06加入反应瓶中,氮气置换,升温至80℃反应8小时。降温至室温,加入乙酸乙酯300ml,水300ml萃取分液,水相再用乙酸乙酯300ml萃取一次;合并有机相,饱和食盐水洗涤,减压浓缩掉有机溶剂,向残留物中加入甲醇100ml,升温至65℃打浆1小时,降温至室温,过滤、干燥,得到化合物vi 17.32g(hplc纯度99.36%,收率82.0%)。ms
+
[m+1]477.2。
[0172]1h nmr(400mhz,dmso-d6)δ12.39(s,1h),8.38(t,j=5.4hz,1h),7.90(s,1h),7.54(s,1h),7.06(dd,j=7.2,1.2hz,1h),6.99(d,j=1.7hz,1h),6.86(td,j=7.7,1.3hz,1h),6.60(td,j=7.4,1.0hz,1h),6.31(d,j=7.8hz,1h),3.85(s,3h),3.76(t,j=8.5hz,2h),
3.47(s,3h),3.26(qd,j=7.3,5.3hz,2h),2.98(t,j=8.4hz,2h),1.14
–
1.06(m,3h).
[0173]
13
c nmr(101mhz,dmso-d6)δ161.49,159.16,154.02,147.82,139.79,133.90,131.99,130.76,129.66,129.09,127.79,126.79,124.60,124.15,118.53,107.93,106.14,104.64,52.99,52.39,35.88,33.73,28.07,14.52.
[0174]
实施例17化合物vi n-乙基-4-[5-甲氧基羰基-3-(吲哚啉-1-基)噻吩-2-基]-6-甲基-7-氧代-6,7-二氢-1h-吡咯并[2,3-c]吡啶-2-甲酰胺的制备
[0175]
在室温下,将1,4-二氧六环40.5ml、水4.5ml、化合物iv 1.5g、化合物v 2.2g、碳酸钾1.83g和四(三苯基膦)钯0.31g加入反应瓶中,氮气置换,升温至80℃反应8小时。降温至室温,加入乙酸乙酯30ml,水30ml萃取分液,水相再用乙酸乙酯30ml萃取一次。合并有机相,饱和食盐水洗涤,减压浓缩掉有机溶剂,向残留物中加入甲醇10ml,升温至65℃打浆1小时,降温至室温,过滤、干燥,得到化合物vi 1.80g(hplc纯度99.68%,收率85.4%)。实施例18化合物vi n-乙基-4-[5-甲氧基羰基-3-(吲哚啉-1-基)噻吩-2-基]-6-甲基-7-氧代-6,7-二氢-1h-吡咯并[2,3-c]吡啶-2-甲酰胺的制备
[0176]
在室温下,将1,4-二氧六环40.5ml、水4.5ml、化合物iv 1.5g、化合物v 2.2g、碳酸钾1.83g、醋酸钯0.06g和三苯基膦0.28g加入反应瓶中,氮气置换,升温至80℃反应8小时。降温至室温,加入乙酸乙酯30ml,水30ml萃取分液,水相再用乙酸乙酯30ml萃取一次。合并有机相,饱和食盐水洗涤,减压浓缩掉有机溶剂,向残留物中加入甲醇10ml,升温至65℃打浆1小时,降温至室温,过滤、干燥,得到化合物vi 1.72g(hplc纯度98.74%,收率81.5%)。实施例19化合物vi n-乙基-4-[5-甲氧基羰基-3-(吲哚啉-1-基)噻吩-2-基]-6-甲基-7-氧代-6,7-二氢-1h-吡咯并[2,3-c]吡啶-2-甲酰胺的制备
[0177]
在室温下,将1,4-二氧六环40.5ml、水4.5ml、化合物iv 1.5g、化合物v 2.2g、碳酸钾1.22g和四(三苯基膦)钯0.31g加入反应瓶中,氮气置换,升温至80℃反应8小时。降温至室温,加入乙酸乙酯30ml,水30ml萃取分液,水相再用乙酸乙酯30ml萃取一次。合并有机相,饱和食盐水洗涤,减压浓缩掉有机溶剂,向残留物中加入甲醇10ml,升温至65℃打浆1小时,降温至室温,过滤、干燥,得到化合物vi 1.62g(hplc纯度99.15%,收率76.8%)。实施例20化合物vi n-乙基-4-[5-甲氧基羰基-3-(吲哚啉-1-基)噻吩-2-基]-6-甲基-7-氧代-6,7-二氢-1h-吡咯并[2,3-c]吡啶-2-甲酰胺的制备
[0178]
在室温下,将1,4-二氧六环40.5ml、水4.5ml、化合物iv 1.5g、化合物v 2.2g、碳酸钾2.14g和四(三苯基膦)钯0.31g加入反应瓶中,氮气置换,升温至80℃反应8小时。降温至室温,加入乙酸乙酯30ml,水30ml萃取分液,水相再用乙酸乙酯30ml萃取一次。合并有机相,饱和食盐水洗涤,减压浓缩掉有机溶剂,向残留物中加入甲醇10ml,升温至65℃打浆1小时,降温至室温,过滤、干燥,得到化合物vi 1.82g(hplc纯度99.03%,收率86.3%)。实施例21化合物vi n-乙基-4-[5-甲氧基羰基-3-(吲哚啉-1-基)噻吩-2-基]-6-甲基-7-氧代-6,7-二氢-1h-吡咯并[2,3-c]吡啶-2-甲酰胺的制备
[0179]
在室温下,将n,n-二甲基乙酰胺40.5ml、水4.5ml、化合物iv 1.5g、化合物v 2.2g、碳酸钾1.83g和四(三苯基膦)钯0.31g加入反应瓶中,氮气置换,升温至80℃反应8小时。降温至室温,加入乙酸乙酯30ml,水30ml萃取分液,水相再用乙酸乙酯30ml萃取一次。合并有机相,饱和食盐水洗涤,减压浓缩掉有机溶剂,向残留物中加入甲醇10ml,升温至65℃打浆1小时,降温至室温,过滤、干燥,得到化合物vi 1.57g(hplc纯度99.72%,收率74.4%)。实施
2-基)-6-甲基-7-氧代-6,7-二氢-1h-吡咯并[2,3-c]吡啶-2-甲酰胺)的制备
[0188]
在室温、惰性气体保护下,将四氢呋喃60ml、化合物vi 2.0g加入反应瓶中,降温至10℃滴加3m甲基溴化镁5.9ml,反应30min。加入饱和氯化铵淬灭反应,调ph至8-9,再加入水12ml,分液,有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩掉有机溶剂,向残留物中加入二氯甲烷16ml和1,4-二氧六环3ml,20℃析晶6小时,过滤、干燥,得到化合物vii 1.48g(hplc纯度98.56%,收率74.0%)。
[0189]
实施例26化合物vii(n-乙基-4-(5-(2-羟基丙基-2-基)-3-(吲哚啉-1-基)噻吩-2-基)-6-甲基-7-氧代-6,7-二氢-1h-吡咯并[2,3-c]吡啶-2-甲酰胺)的制备
[0190]
在室温、惰性气体保护下,将四氢呋喃60ml、化合物vi 2.0g加入反应瓶中,降温至10℃滴加1.6m甲基锂11.8ml,反应30min。加入饱和氯化铵淬灭反应,调ph至8-9,再加入水12ml,分液,有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩掉有机溶剂,向残留物中加入二氯甲烷16ml和1,4-二氧六环3ml,20℃析晶6小时,过滤、干燥,得到化合物vii 1.46g(hplc纯度97.21%,收率73.0%)。
[0191]
实施例27化合物vii(n-乙基-4-(5-(2-羟基丙基-2-基)-3-(吲哚啉-1-基)噻吩-2-基)-6-甲基-7-氧代-6,7-二氢-1h-吡咯并[2,3-c]吡啶-2-甲酰胺)的制备
[0192]
在室温、惰性气体保护下,将2-甲基四氢呋喃60ml、化合物vi 2.0g加入反应瓶中,降温至10℃滴加1.6m甲基锂11.1ml,反应30min。加入饱和氯化铵淬灭反应,调ph至8-9,再加入水12ml,分液,有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩掉有机溶剂,向残留物中加入二氯甲烷16ml和1,4-二氧六环3ml,20℃析晶6小时,过滤、干燥,得到化合物vii 1.55g(hplc纯度97.94%,收率77.5%)。
[0193]
实施例28化合物vii(n-乙基-4-(5-(2-羟基丙基-2-基)-3-(吲哚啉-1-基)噻吩-2-基)-6-甲基-7-氧代-6,7-二氢-1h-吡咯并[2,3-c]吡啶-2-甲酰胺)的制备
[0194]
在室温、惰性气体保护下,将四氢呋喃60ml、化合物vi 2.0g加入反应瓶中,降温至20℃滴加1.6m甲基锂11.1ml,反应30min。加入饱和氯化铵淬灭反应,调ph至8-9,再加入水120ml,分液,有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压浓缩掉有机溶剂,向残留物中加入二氯甲烷16ml和1,4-二氧六环3ml,20℃析晶6小时,过滤、干燥,得到化合物vii 1.49g(hplc纯度98.99%,收率74.5%)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1