一种嗜氮酮类化合物及其制备方法和应用
1.本发明属于海洋天然药物领域,具体涉及两个嗜氮酮类化合物及其制备方法和应用。
背景技术:
2.嗜氮酮类化合物属于聚酮类化合物,是一类结构较为多样的真菌聚酮化合物,具有高度氧化的吡喃醌双环核(通常称为异戊二烯)和季碳中心,一般为红色或者是黄色,属于天然的真菌色素,是一种天然理想的可用于食品的着色剂;同时它具有良好的细胞毒性、抗病毒、抗微生物以及抗炎等生物活性,具有开发药物先导化合物的巨大潜力。
3.菌核青霉可以产生出结构多种多样的次级代谢产物,其中嗜氮酮类化合物是一类结构较为多样、具有显著优异生物活性的次级代谢产物,海洋真菌可以产生结构独特的次级代谢产物,是药物先导化合物的重要来源,已经成为了嗜氮酮类化合物的一种主要且重要的来源。
技术实现要素:
4.本发明提供两个嗜氮酮类化合物,其结构式如式(i)所示:式(i)。
5.本发明的另一个目的是提供一种上述嗜氮酮类化合物的制备方法。
6.为实现上述目的,本发明采用如下技术方案。
7.一种菌核青霉(penicillium sclerotiorum)ujnmf 0503,保藏于中国微生物菌种保藏管理委员会普通微生物中心(cgmcc),保藏号:cgmcc no. 40172。
8.一种菌核青霉(penicillium sclerotiorum) ujnmf 0503及其发酵产物在生产具有抗菌、抗氧化、α-糖苷酶抑制剂及其应用。所述菌核青霉(penicillium sclerotiorum)ujnmf 0503培养后可产生发酵产物,所述发酵产物中含有抗菌、抗氧化、抑制α-糖苷酶作用的化合物,其发酵液可用于生产具有抗菌、抗氧化、α-糖苷酶抑制剂的组合物、药物、保健品或添加剂。
9.一种菌核青霉(penicillium sclerotiorum) ujnmf 0503产生的嗜氮酮类化合物,其结构如式(i)所示。
10.式(i)嗜氮酮类化合物的制备方法,包括以下步骤:(1)菌核青霉(penicillium sclerotiorum)ujnmf 0503的发酵;(2)从步骤(1)所得发酵物中经提取得到发酵提取物;(3)步骤(2)中的提取物通过正相硅胶柱、快速中压层析柱、高效液相色谱等方法
进行分离。
11.本发明提供一种含有步骤(2)中提取物,或式(i)所示化合物在制备抗菌、抗氧化、α-糖苷酶抑制剂等保健品、药物或药物中间体中的用途。
12.本发明提供一种含有步骤(2)中提取物,或式(i)所示化合物的抗菌、抗氧化、抑制α-糖苷酶作用的保健品或药物。所述抗菌、抗氧化、α-糖苷酶抑制剂保健品或药物还包括医学上可接受的辅料。所述抗菌、抗氧化、α-糖苷酶抑制剂的保健品或药物还可以包括其他有效成分,以增强效果或扩大应用范围。
13.本发明具有以下优点:本发明的嗜氮酮类化合物可以通过penicilliumsclerotiorumujnmf0503的发酵提取分离获得,具有抗菌、抗氧化、α-糖苷酶抑制剂,在制备抗菌、抗氧化、α-糖苷酶抑制剂的组合物、药物、保健品或添加剂等方面具有应用潜力。
14.生物保藏信息菌核青霉(penicilliumsclerotiorum)ujnmf0503于2022年05月13日保藏于中国微生物菌种保藏管理委员会普通微生物中心(cgmcc),保藏地址为中国北京,北京市朝阳区北辰西路1号院3号,中国科学院微生物研究所,保藏编号为cgmccno.40172。
附图说明
15.图1为真菌penicilliumsclerotiorumujnmf0503平板培养图片。
16.图2为式(i)所示化合物主要的1h-1
hcosy、hmbc信息。
17.图3为式(i)所示化合物的ecd谱图。
具体实施方式
18.下面结合实施例和附图对本发明做进一步说明,但本发明不受下述实施例的限制。实施例中的实验方法,如无特别说明,均采用本领域常规技术,实验试剂均为商业购买。
19.实施例1发酵物的制备种子培养基的配制方法:土豆浸出液200ml,葡萄糖20g,海盐30g,用水定容到1l。将培养基装入20个500ml的三角烧瓶中,每瓶约150ml,在121℃高压蒸汽灭菌25分钟,备用。
20.pdb发酵培养基配置方法:土豆浸出液200ml,葡萄糖20g,海盐30g,用水定容到1l。置于1l三角瓶中,每瓶350ml液体培养基,共150瓶。121℃高压蒸汽灭菌25分钟,备用。
21.用无菌竹签挑取适量的真菌penicilliumsclerotiorumujnmf0503菌种接种入种子培养基中,28℃摇床(160rpm)培养3天得到种子液,然后用移液枪接种10ml种子液到1l的装有pdb培养基的三角烧瓶中,于28℃静置培养30天后,收取发酵物。
22.将发酵物用纱布进行过滤分为菌体和菌液,菌体用95%乙醇浸泡,浸取液回收乙醇后剩余水相,再用乙酸乙酯进行萃取,减压浓缩得到乙酸乙酯提取物a,菌液用等量乙酸乙酯萃取三次,减压浓缩得到乙酸乙酯提取物b,a、b合并得到总提取物。
23.实施例2嗜氮酮类化合物的制备按照实施例1的方法获得70l液体培养基,菌体用95%乙醇浸泡、浸取液回收乙醇后剩余水相再用乙酸乙酯进行萃取,减压浓缩得到乙酸乙酯提取物a,菌液用等量乙酸乙酯
萃取三次,减压浓缩得到乙酸乙酯提取物b,a、b合并得到总提取物200g。乙酸乙酯提取物用大孔树脂吸附材料进行柱层析,以乙醇/水作为洗脱剂,体积比30%、50%、75%、90%进行洗脱,回收洗脱溶剂,获得4个馏分(fr.1-fr.4)。
24.fr.3通过硅胶柱层析(100-200目),以二氯甲烷/甲醇(100:0、90:10、80:20、70:30、50:50、0:100)进行梯度洗脱,结合薄层色谱硅胶板检测,共得到8个馏分(fr.3.1-fr.3.8)。fr.3.2通过硅胶柱层析(200-300目),以石油醚/乙酸乙酯(100:0、90:10、80:20、70:30、50:50、0:100)进行梯度洗脱,合并得到4个馏分(fr.3.2.1-fr.3.2.4),fr.3.2.3通过半制备高效液相色谱(色谱柱为chiralmz(2)rh5u10mm
×
250mm,流速为3.0ml/min,检测波长280nm和320nm,流动相为体积比为80:20的甲醇-水)得到式i所示化合物1(tr=15.4min,2.2mg)和2(tr=16.8min,2.6mg)。
25.式(i)所示化合物均为红褐色胶状物,易溶于氯仿、甲醇、dmso,难溶于水,旋光度值分别为[α]
27d
+8.7(c0.24,meoh)、[α]
27d
–
183.0(c0.15,meoh)。
[0026]
对分离获得的化合物进行高分辨质谱(hr-esims)、1hnmr、
13
cnmr、2d1h-1
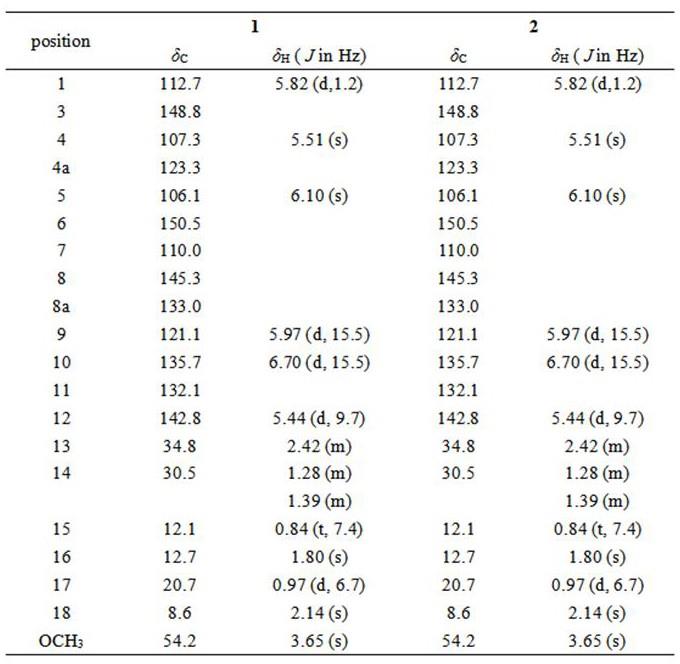
hcosy、hsqc、hmbc分析,确定了平面结构,再结合生源和ecd谱图确定了化合物的绝对构型。化合物的1h和
13
cnmr数据见表1,化合物1主要的1h-1
hcosy和hmbc相关信息见图2,ecd谱图见图3。
[0027]
表1化合物1-2在cdcl3中的1h和
13
cnmr数据
结构鉴定:化合物1:红褐色胶状,高分辨质谱(hresims)在m/z处给出准分子离子峰347.1852([m + h] +
,计算347.1853),结合nmr数据,推测化合物的分子式为c
20h27
o5,不饱和度为8。hsqc和dept谱数据显示该化合物含有7个sp2季碳,5个sp2次甲基,2个次甲基,1个亚甲基,4个甲基和1个甲氧基;1h nmr数据显示,该化合物含有4个甲基δ
h 0.84 (t, 7.4, h
3-15),1.80 (s, h
3-16),0.97 (d, 6.7, h
3-17),2.14 (s, h
3-18),1个亚甲基 δ
h 1.39 (m, h-14a),1.28 (m, h-14b),5个sp2次甲基质子δh5.51 (s, h-4),6.10 (s, h-5),5.97 (d, 15.5, h-9), 6.70 (d, 15.5, h-10),5.44 (d, 9.7, h-12),1个sp3次甲基质子δh2.42 (m, h-13)。1h-1
h cosy谱中h-9与h-10相关,h-13和h-12、h-14、h
3-17分别相关, h-14与h-15相关,hmbc谱中h
3-16和c-10、c-11、c-12相关,可以推测出c9-c17的3,5-dimethyl-1,3-heptadiene片段,不饱和度为2。余下部分(c
11h11
o5)高度不饱和,推测含有苯环。hmbc谱中,6-oh与c-5、c-6、c-7相关,h
3-18与c-6、c-7、c-8、c-8a相关,8-oh与c-6、c-7、c-8、c-4a相关,h-1与c-3、c-8a相关,h-4与c-3、c-5、c-4a相关,och3与c-1相关,可以推测该化合物含有c1-c8的结构片段,最后通过hmbc谱中h-9与c-3、c-4的相关信号将两个片段连接起来。
[0028]
化合物2:红褐色胶状,高分辨质谱(hresims)在m/z处给出准分子离子峰347.1856([m + h] +
,计算347.1853),结合nmr数据,推测化合物的分子式为c
20h27
o5,不饱和度为8。
nmr分析发现化合物2与化合物1基本一致,推测两者为差相异构体。
[0029]
通过计算与实验ecd比较,推测两化合物c-1位绝对构型分别为r和s。
[0030]
化合物的结构式如下:实施例3式(i)所示化合物的抗菌活性测试选择staphylococcusaureusatcc25923作为测试菌株,首先将菌株接种到lb固体培养基上进行复苏,然后挑取适量菌落接种到液体培养基37℃、169rpm摇床震荡培养,使用空白培养基将培养好的菌株稀释到105cfu。使用移液枪量取190μl加入至96孔板中,再加入10μl含有一定浓度的化合物的dmso溶液,37℃过夜培养,使用酶标仪在600nm下测定吸光度(od值)并计算抑制率,实验使用空白培养基作为阳性对照。
[0031]
实验结果显示,在测试浓度为50μm时,式(i)所示化合物1和2对金黄色葡萄球菌的抑制率分别为97.5%和98.1%,ic
50
值为28.36μm和32.17μm。表明化合物1和2对金黄色葡萄球菌具有一定的抑制作用。
[0032]
实施例4dpph法测定式(i)所示化合物的抗氧化活性无水乙醇配制dpph乙醇溶液备用,乙醇配制一定浓度的样品备用。使用移液枪量取50μldpph乙醇溶液和50μl样品加入96孔板中,量取100μl乙醇加入96孔板中作为空白对照a0,量取50μldpph乙醇溶液和50μl姜黄素加入到96孔板中作为阳性对照ac,使用酶标仪在25℃下孵育30min并测量其在517nm下的吸光度ai,计算抑制率。
[0033]
实验结果显示,式(i)所示化合物1和2清除dpph氧自由基的ic
50
值分别为44.54μm和36.83μm,姜黄素作为阳性对照,其ic
50
值为33.42μm,表明化合物1和2具有良好的抗氧化活性实施例5式(i)所示化合物抑制α-糖苷酶的活性测试96孔板每孔加入99μl的pbs磷酸缓冲液(ph=6.8),然后将20mm的1μl待测化合物溶液或空白对照加入到对应的孔中,随后加入25μl的α-葡萄糖苷酶溶液,37℃摇床孵育15min。加入25μl的pnpg溶液,再置于37℃摇床孵育15min,随后加入50μl的0.2m碳酸钠溶液,酶标仪测定405nm处的吸光度,计算待测化合物对α-葡萄糖苷酶的抑制率。
[0034]
实验结果显示,式(i)所示化合物1和2有一定的抑制α-糖苷酶活性,50μm的化合物抑制率分别为42.1%和57.0%,阳性对照药阿卡波糖抑制率为75.2%,说明化合物具有开发为α-糖苷酶抑制剂药物、保健品或添加剂的潜力。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1