嘧啶氨基吡唑衍生物及其作为富亮氨酸重复激酶2抑制剂的应用
1.本发明属于化学药物技术领域,涉及嘧啶氨基吡唑衍生物及其作为富亮氨酸重复激酶2抑制剂的应用。
背景技术:
2.帕金森氏病(pd)是一种高发于中老年的第二大慢性神经退行性疾病,仅次于阿尔兹海默症。自从1817年被发现命名以来,人类与它抗争的历史已有二百多年。目前人们对该疾病认知度低、就诊率低、诊断率低,且无法治愈,患者终身表现为震颤、肢体僵硬、运动功能减退和步态异常等运动神经系统障碍,以及嗅觉减退、睡眠障碍、便秘等非运动症状。现有的药物,仅能针对症状本身进行不同程度的缓解,无法控制疾病进展。目前的临床常用药物,远远不能满足现有帕金森中晚期患者的需求,亟需一类可防止帕金森病理、生化退行的药物。疾病修饰治疗是目前开发帕金森疾病治疗药物的主流方向,可以影响神经元变性的初始触发因素,并促使神经元代偿反应或减少病理传播和进展。目前主流研究认为,路易小体(lb)内α-syn聚集是帕金森病发病的重要原因,减少α-syn的聚集是治疗帕金森病的潜在方法。
3.而富亮氨酸重复激酶2(leucine-rich repeat kinase 2,lrrk2)阻断分子伴侣介导自噬,导致α-syn不能被降解,产生毒性。lrrk2参与α-syn介导的神经毒性,lrrk2通过氧化机制诱导线粒体损伤、内溶酶体功能障碍,诱发帕金森病进展。lrrk2激酶抑制剂可以减轻帕金森疾病模型的病理性损害,改善患者运动功能障碍。两种新型lrrk2激酶抑制剂dnl201和dnl151的安全性和耐受性ib期临床试验获得成功,dnl151已经开展iib/iii期注册临床研究。因此lrrk2小分子抑制剂的开发,是目前最有潜力开发帕金森治疗药物的研究方向之一。
4.因此,开发有效的lrrk2激酶以及突变的lrrk2激酶的抑制剂成为目前治疗神经退行性疾病的一条重要的途径。本发明旨在发明一种可以高度对lrrk2激酶抑制的化合物,从而进一步发明可以很好的治疗神经退行性疾病的药物。
5.专利us8802674b2公开了gentech公司嘧啶氨基苯酰胺类化合物是一类lrrk2的抑制剂,其化学结构式如下所示:
[0006][0007]
us09212173b2公开了gentech公司嘧啶氨基吡唑类化合物是一类lrrk2的抑制剂,
其化学结构式如下:
[0008][0009]
在该公司后期的研究中发现,上述类别化合物在动物实验中都发现对周边组织(比如肾脏和肺)有潜在的毒副作用。鉴于帕金森类疾病近些年还没有1个药物研究成功,临床上只有1个新药在临床研究。因此,更多高活性的新结构小分子lrrk2激酶抑制剂有待被发现。
技术实现要素:
[0010]
有鉴于此,本发明的目的在于提供一种嘧啶氨基吡唑衍生物及其作为富亮氨酸重复激酶2抑制剂的应用。
[0011]
为达到上述目的,本发明提供如下技术方案:
[0012]
1、嘧啶氨基吡唑衍生物,或其光学异构体,或其前体药物,或其药学上可接受的盐,或其水合物、溶剂化物、n-氧化物、氘代物,该衍生物的通式结构如下:
[0013][0014]
其中,r1选自h、f、cl、br或i;
[0015]
r2选自h、f、cl、br或i;
[0016]
而且n和m分别选自1,2,3。
[0017]
r3选自分别为h、c
1-6
烷基、氧代的c
1-6
烷基或者卤代的c
1-6
烷基。。
[0018]
优选的,所述衍生物为
[0019]n2-(5-氯-1-(1-(2-氟代乙酯)哌啶-4-基)-1h-吡唑-4-基)-n
4-甲基-5-(三氟甲基)嘧啶-2,4-联胺,n
2-(5-chloro-1-(1-(2-fluoroethyl)piperidin-4-yl)-1h-pyrazol-4-yl)-n
4-methyl-5-(trifluoromethyl)py rimidine-2,4-diamine,其化学结构式如下:
[0020][0021]
优选的,所述衍生物为
[0022]n2-(3-氯-1-(1-(2-氟代乙酯)哌啶-4-基)-1h-吡唑-4-基)-n
4-甲基-5-(三氟甲基)嘧啶-2,4-联胺,n
2-(3-chloro-1-(1-(2-fluoroethyl)piperidin-4-yl)-1h-pyrazol-4-yl)-n
4-methyl-5-(trifluoromethyl)py rimidine-2,4-diamine,其化学结构式如下:
[0023][0024]
2、嘧啶氨基吡唑衍生物,或其光学异构体,或其前体药物,或其药学上可接受的盐,或其水合物、溶剂化物、n-氧化物、氘代物作为富亮氨酸重复激酶2抑制剂的应用。
[0025]
3、嘧啶氨基吡唑衍生物,或其光学异构体,或其前体药物,或其药学上可接受的盐,或其水合物、溶剂化物、n-氧化物、氘代物在制备治疗或预防帕金森病药物中的应用。
[0026]
4、嘧啶氨基吡唑衍生物,或其光学异构体,或其前体药物,或其药学上可接受的盐,或其水合物、溶剂化物、n-氧化物、氘代物在制备治疗或预防慢性神经退行性疾病药物中的应用。
[0027]
5、嘧啶氨基吡唑衍生物,或其光学异构体,或其前体药物,或其药学上可接受的盐,或其水合物、溶剂化物、n-氧化物、氘代物在制备抑制富亮氨酸重复激酶2的活性而预防和/或治疗疾病的药物中的应用。
[0028]
6、一种药物组合物或制剂,包含前述嘧啶氨基吡唑衍生物,或其光学异构体,或其前体药物,或其药学上可接受的盐,或其水合物、溶剂化物、n-氧化物、氘代物。
[0029]
优选的,还包含药学上可接受的辅料、辅助剂或载体。
[0030]
本发明的有益效果在于:
[0031]
申请人在嘧啶氨基吡唑为骨架的基础上进一步发现了一类含氮杂环衍生物,相比现有类似骨架结构化合物,该衍生物显示出对lrrk2活性更强的抑制作用。是一类潜在的中枢神经疾病(如:帕金森)治疗效应的化合物。
附图说明
[0032]
为了使本发明的目的、技术方案和有益效果更加清楚,本发明提供如下附图进行说明。
[0033]
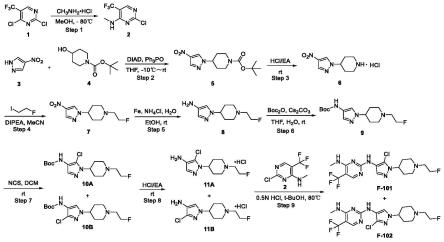
图1为实施例1中f101、f102的合成路线。
具体实施方式
[0034]
下面将结合附图,对本发明的优选实施例进行详细的描述。
[0035]
定义和说明
[0036]
除非另有说明,本文所用的下列术语和短语旨在具有下列含义。一个特定的术语或短语在没有特别定义的情况下不应该被认为是不确定的或不清楚的,而应该按照普通的含义去理解。当本文中出现商品名时,意在指代其对应的商品或其活性成分。
[0037]
这里所采用的术语“药学上可接受的”,是针对那些化合物、材料、组合物和/或剂型而言,它们在可靠的医学判断的范围之内,适用于与人类和动物的组织接触使用,而没有过多的毒性、刺激性、过敏性反应或其它问题或并发症,与合理的利益/风险比相称。
[0038]
术语“药学上可接受的盐”是指本发明化合物的盐,由本发明发现的具有特定取代基的化合物与相对无毒的酸或碱制备。当本发明的化合物中含有相对酸性的功能团时,可以通过在纯的溶液或合适的惰性溶剂中用足够量的碱与这类化合物的中性形式接触的方式获得碱加成盐。药学上可接受的碱加成盐包括钠、钾、钙、铵、有机胺或镁盐或类似的盐。
当本发明的化合物中含有相对碱性的官能团时,可以通过在纯的溶液或合适的惰性溶剂中用足够量的酸与这类化合物的中性形式接触的方式获得酸加成盐。本发明的某些特定的化合物含有碱性和酸性的官能团,从而可以被转换成任一碱或酸加成盐。
[0039]
本发明的药学上可接受的盐可由含有酸根或碱基的母体化合物通过常规化学方法合成。一般情况下,这样的盐的制备方法是:在水或有机溶剂或两者的混合物中,经由游离酸或碱形式的这些化合物与化学计量的适当的碱或酸反应来制备。
[0040]
术语“有效治疗剂量”指的是在给予需要这样的治疗的哺乳动物时,足以有效治疗的通式化合物的量。治疗有效量将依赖于所用的治疗药剂的特定活性、患者的年龄、生理状况、其它疾病状态的存在和营养状况而变化。此外,患者可能正接受的其它药物治疗将影响要给予的治疗药剂的治疗有效量的确定。
[0041]
术语“治疗”意味着对于哺乳动物体内疾病的任何治疗,包括:(i)防止疾病,即造成疾病的临床症状不发展;(ii)抑制疾病,即阻止临床症状的发展;和/或(iii)减轻疾病,即造成临床症状的消退。
[0042]
术语“药学上可接受的辅料、辅助剂或载体”包括任何和全部的溶剂、分散介质、包衣、抗细菌和抗真菌药剂、等渗和吸收延迟剂等。这样的介质和药剂用于药学活性物质在本领域是众所周知的。除非任何常规介质或药剂与活性成分不相容,其在治疗组合物中的应用是可预期的。补充的活性成分也可以并入组合物中。
[0043]
本发明的化合物可以通过本领域技术人员所熟知的多种合成方法来制备,包括下面列举的具体实施方式、其与其他化学合成方法的结合所形成的实施方式以及本领域技术上人员所熟知的等同替换方式,优选的实施方式包括但不限于本发明的实施例。凡是通过其他反应条件能得到合成路线中的中间体或者化合物1均视为该发明的替代方案。
[0044]
实施例1:目标化合物f-101和f-102的合成
[0045]
合成路线如图1所示。
[0046]
具体步骤如下:
[0047]
step 1:中间体2的制备
[0048]
将2,4-二氯-5-三氟甲基嘧啶(5.00g,23.04mmol),三乙胺(4.66g,46.08mmol)加入到50.0ml甲醇中,降温至-80℃。降温完毕后将甲胺盐酸盐(1.56g,23.04mmol)分批加入到反应体系中,加毕,-80℃反应2h。tlc监测反应进程,反应完成后升温至室温。减压浓缩除去溶剂,得到粗产品。粗产物通过硅胶柱层析纯化(石油醚/乙酸乙酯=10/1,体积比),得到中间体2(1.34g,27.5%,白色固体),[m+h]
+
=212。
[0049]
step 2:中间体5的制备
[0050]
将中间体3(2.26g,20.00mmol),中间体4(4.83g,24.00mmol)和三苯基膦(9.44g,36.00mmol)加入到20.0ml四氢呋喃中,搅拌溶清,降温至低于-10℃。称取偶氮二羧酸二异丙酯(7.28g,36.00mmol)溶于10ml四氢呋喃中,缓慢滴入上述反应瓶中,滴毕,缓慢升至室温反应17h。lc-ms和tlc监测,原料反应完全。向反应液中加入200.0ml水,用乙酸乙酯萃取两次,合并乙酸乙酯相,无水硫酸钠干燥,过滤,浓缩,得粗产物中间体5(2.50g,43.0%,黄色油状物),[m+h]
+
=297,[m-boc+h]
+
=197。直接投入下一步。
[0051]
step 3:中间体6的制备
[0052]
将中间体5(25.50g,86.10mmol)溶于乙酸乙酯中,加入2m盐酸乙酸乙酯溶液
(50.0ml),加毕,室温搅拌3h。lc-ms监测原料未反应完,补加2m盐酸乙酸乙酯溶液(20.0ml)室温搅拌1h,lc-ms监测原料反应完。过滤,滤饼油泵冷抽至干,得到中间体6(3.26g,16.3%,类白色固体),[m+h]
+
=197。
[0053]
step 4:中间体7的制备
[0054]
向中间体6(1.62g,8.25mmol)和碳酸铯(10.70g,33.00mmol)的n,n-二甲基甲酰胺(20.0ml)溶液中加入1-氟-2-碘乙烷(1.72g,9.90mmol),加毕,升温至50℃反应2.5h。lc-ms监测,原料反应完全。加入150.0ml水,乙酸乙酯萃取,萃取的有机相经饱和氯化钠洗涤,无水硫酸钠干燥,过滤,浓缩,得粗产物中间体7(2.03g,120.0%,黄色油状物),[m+h]
+
=243。直接投入下一步。
[0055]
step 5:中间体8的制备
[0056]
取反应试管,加入中间体7(2.03g,8.30mmol),乙醇10.0ml,铁粉(1.40g,25.00mmol)和饱和氯化铵水溶液10.0ml,室温反应0.5h。lc-ms监测原料大部分未反应,补加铁粉(1.00g,17.80mmol),室温搅拌45min。lc-ms监测原料反应完。硅藻土过滤,滤饼乙醇淋洗,将乙醇浓缩至干,向残余物中加入二氯甲烷萃取,萃取的有机相经无水硫酸钠干燥,过滤,浓缩,得中间体8(1.93g,108.5%,棕黑色油状物),[m+h]
+
=213。直接投入下一步。
[0057]
step 6:中间体9的制备
[0058]
向中间体8(1.93g,9.10mmol)的四氢呋喃(10.0ml)和水(10.0ml)的混合液中依次加入碳酸铯(5.54g,17.00mmol)和boc2o(2.10g,9.60mmol),室温反应1h。lc-ms监测原料反应完,反应液使用乙酸乙酯萃取。萃取的有机相经无水硫酸钠干燥,过滤,浓缩,得紫色油状物3.20g,粗产物通过硅胶柱层析纯化,得到中间体9(970.0mg,34.1%,固体),[m+h]
+
=313,[m+h-100]
+
=213,[m+h-56]
+
=257。
[0059]
step 7:中间体10a和10b的制备
[0060]
取反应试管,加入中间体9(800.0mg,2.56mmol),ncs(376.0mg,2.82mmol),二氯甲烷16.0ml,室温反应18h。lc-ms和tlc监测原料反应约一半,停止反应,反应液经硫代硫酸钠洗涤,水洗,无水硫酸钠,过滤,浓缩,得粗产物。粗产物通过硅胶柱层析纯化,得到中间体10a和10b(10a:174.0mg,19.6%;10b:154.0mg,17.3%),分别浓缩,直接投入下一步。[m+h]
+
=347,[m+h-100]
+
=247,[m+h-56]
+
=291。
[0061]
step 8:中间体11a和11b的制备
[0062]
将中间体10a(174.0mg,0.50mmol)加入到4.0ml盐酸乙酸乙酯溶液(2m),室温反应14h,lc-ms监测原料反应完。反应液过滤,乙酸乙酯淋洗,快速收集固体(固体易吸潮),油泵冷抽至干,得中间体11a(134.0mg,94.3%,类白色固体),[m+h]
+
=247。经同样的方法得到11b。
[0063]
step 9:化合物f-101和f-102的制备
[0064]
取反应试管,加入中间体11a(134.0mg,0.47mmol),中间体2(150.0mg,0.71mmol),醋酸(14.0mg,0.24mmol)和t-buoh(7.0ml),升温至50℃反应过夜。lc-ms监测少量原料未反应完,停止反应。将反应液过滤,乙酸乙酯淋洗得到固体产物。将固体用二氯甲烷溶解,加入饱和碳酸氢钠洗涤,分液,水相二氯甲烷提取两次,合并二氯甲烷相。合并的有机相经无水硫酸钠干燥,过滤,浓缩得到粗产物。粗产物通过硅胶柱层析纯化(二氯甲烷/甲醇=10/1,体积比),干燥得到终产物(f-101:102.0mg,51.1%,类白色固体,[m+h]
+
=422);1h nmr
(400mhz,dmso-d6)δ9.00(s,1h),8.08(s,1h),7.84(s,1h),7.03(s,1h),4.60(t,j=4.9hz,1h),4.48(t,j=4.9hz,1h),4.28
–
4.15(m,1h),3.00(d,j=11.8hz,2h),2.82(s,3h),2.69(t,j=4.9hz,1h),2.62(t,j=4.9hz,1h),2.26
–
2.15(m,2h),2.08
–
1.93(m,2h),1.89
–
[0065]
1.77(m,2h)。经相同的方法,由中间体11b得到f-102:97.0mg,42.2%,类白色固体),[m+h]
+
=422。1h nmr(400mhz,dmso-d6)δ9.00(s,1h),8.08(s,1h),7.84(s,1h),7.03(s,1h),4.60(t,j=4.9hz,1h),4.48(t,j=4.9hz,1h),4.28
–
4.15(m,1h),3.00(d,j=12.2,2h),2.82(s,3h),2.69(t,j=4.9hz,1h),2.62(t,j=4.9hz,1h),2.20(td,j=11.9,2.3hz,2h),2.01(qd,j=12.2,3.8hz,2h),1.87
–
1.78(m,2h)。
[0066]
生物活性测试
[0067]
蛋白结合实验:
[0068]
试剂耗材:
[0069]
lrrk2 g2019s酶(赛默飞),底物(lrrktide)(赛默飞),atp(赛默飞)tr-fret稀释液(赛默飞),plrrktide抗体(赛默飞),384孔板(pe)dmso(索莱宝)
[0070]
实验过程:
[0071]
所有待测化合物(包括阳性对照和待测样品)用dmso稀释至1mm,得到相应的测试化合物溶液。35μl阳性化合物(结构式见表1,genentech公司在文献(bryan k,chan,anthony a,et al.discovery of a highly selective,brain-penetrant aminopyrazole lrrk2 inhibitor.[j].acs med chem lett.2012nov 23;4(1):85-90.中公开的类似结构化合物,并参照该文献合成)溶液、35μl测试化合物溶液、35μl空白溶液次加入384孔板中,将板在2500rpm下离心1分钟以1mm为初始浓度,3倍梯度稀释10个点,并且按照每孔100nl阳性化合物、测试化合物、空白孔溶液分别加入至另一块384测定板中,3个复孔,将板在2500rpm下离心1分钟并且在箔中密封待用。
[0072]
酶反应:用测定缓冲液(赛默飞tr—fret dilution buffer)稀释lrrktide底物和lrrk2g2019s激酶混合工作液(终浓度为lrrktide底物:400nm和lrrk2 g2019s激酶:580ng/ml)加入到上述384测定板的所有样品孔中,每孔5μl,384测定板在23℃下孵育20分钟。孵育后,用测定缓冲液稀释2
×
atp工作液(134μm)加入到每个孔中,每孔5ml,384测定板23℃下孵育60分钟。
[0073]
检测:用测定缓冲液(tr-fret dilution buffer)稀释edta和plrrktide抗体得到混合工作液(终浓度为edta:10mm,plrrktide抗体:2nm)。随后向上述384测定板每孔中加入10μl该抗体混合工作液,23℃下孵育60分钟。340nm激发光,520nm荧光发射光和490nm铽发射光的te-fret模式中在酶标仪读板。
[0074]
方法参考:j德比森特菲达尔戈等.化合物、组合物和方法:cn113939294a[p].2022-01-14。
[0075]
各化合物的活性数据见表1。
[0076]
表1.化合物活性数据表
[0077][0078]
由表1可知,本发明实施例所提供的系列新化合物,对于激酶lrrk2 g2019s有较强的抑制作用,明显强于阳性对照(genentech公司在文献(bryan k,chan,anthony a,et al.discovery of a highly selective,brain-penetrant aminopyrazole lrrk2 inhibitor.[j].acs med chem lett.2012nov 23;4(1):85-90.中公开的类似结构化合物)对lrrk2 g2019s的抑制活性。在制备预防和/或治疗体内基因lrrk2活性升高有关的疾病的药物中具有潜在的应用价值。
[0079]
最后说明的是,以上优选实施例仅用以说明本发明的技术方案而非限制,尽管通过上述优选实施例已经对本发明进行了详细的描述,但本领域技术人员应当理解,可以在形式上和细节上对其作出各种各样的改变,而不偏离本发明权利要求书所限定的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1