改性PETG共聚酯、制备方法及其发泡颗粒的制备方法与流程
改性petg共聚酯、制备方法及其发泡颗粒的制备方法
技术领域
1.本发明属于高分子材料及其制备技术领域,具体涉及改性低温耐冲击petg共聚酯、制备方法及其发泡颗粒的制备方法。
背景技术:
2.对苯二甲酸-乙二醇-新戊二醇共聚酯(petg)本身具有透明性高、低温耐冲击性能、耐候性及耐化学性,拥有优秀的加工性能,在板材、片材、热收缩膜以及日化容器、化妆品瓶等领域得到了广泛应用;尤其是用新戊二醇(npg)作为原料合成的petg材料,但是该材料在“浙江理工大学学报”2005年12月22卷第4期中,缪国华,秦伟明,程贞娟在《新戊二醇(npg)对pet的改性研究》的研究中提到在反应过程中,存在着npg与eg的竞聚现象,并且现在“一步法”生产petg的工艺,在连续生产中存在npg含量不稳定的问题。
3.petg作为发泡成型制品少有报道,cn113929886a公开了一种长链支化petg共聚酯及其制备方法,所述共聚酯由一种二元酯、两种二元醇及多官能度支化剂通过酯化缩聚反应制得。支化结构的引入为线性聚酯材料提供了链之间的缠结相互作用,具有更好的熔体强度和撕裂强度,能够显著改善传统线性聚酯存在的熔体强度低等问题,有效拓展了在挤出发泡领域上的应用。但是使用支化剂可能会在聚合过程中粘度逐渐增大,最后失去流动性形成凝胶。
4.《塑料》2012年41卷第3期中,王向东,何路东,刘本刚,周洪福在《petg连续挤出发泡行为》的研究中采用环氧型扩链剂petg进行熔融扩链,扩链剂的加入能有效提高petg树脂的熔体强度和改善其“可发泡性”,在发泡成型过程中可以有效阻止泡孔的塌陷和破裂,进而形成泡孔尺寸和形态分布较为均匀的制品;而使用扩链剂进行扩链会导致petg的分子量分布变宽,材料易发生老化,力学性能也会在低温条件下有所下降。
5.现在发泡工艺应用最为广泛的是超临界co2发泡,此发泡方式具有无毒、不可燃、安全、发泡效率高等优点,且超临界co2对聚合物熔体有很好的的增塑作用,能降低聚合物熔体的黏度,提高熔体的流动性,降低挤出温度。然而用新戊二醇制备的petg具有两个侧位甲基,其空间位阻导致petg分子链对co2亲和性降低,co2溶解度下降,导致使用超临界co2发泡时发泡倍率低、膜泡不稳定。所以目前该材料用超临界co2进行发泡的效果并不是很好。
6.如何解决制备的petg中npg的含量稳定,以及改善材料对co2的溶解度、提升发泡倍率,改善发泡材料性能是现阶段研究的一个重点。
技术实现要素:
7.本发明要解决的技术问题是克服现有技术的不足,提供一种改性petg共聚酯,低温耐冲击性能好,利用其制备的改性petg共聚酯发泡颗粒,发泡倍率高、泡孔均匀;本发明还提供改性petg共聚酯的制备方法和改性petg发泡颗粒的制备方法。
8.本发明所述的改性petg共聚酯的制备方法,包括以下步骤:(1)分酯化反应:将对苯二甲酸、端羧基有机硅氧烷、新戊二醇、乙二醇、催化剂a、
交联剂混合进行酯化反应,得到低聚酯化物a;将对苯二甲酸、端羧基有机硅氧烷、乙二醇、催化剂a、交联剂混合进行酯化反应,得到低聚酯化物b。
9.(2)缩聚反应:将低聚酯化物a和低聚酯化物b混合,加入催化剂a、助剂,进行缩聚反应,得到改性petg共聚酯。
10.催化剂a为钛酸四丁酯、硅酸四乙酯和苯基膦酸、醋酸锑、乙二醇锑中的一种。
11.催化剂b为铂、氯铂酸的异丙醇溶液或者铂(0)-1,3-二乙烯基四甲基二硅氧烷络合物(含pt量19.0-21.5%)。
12.交联剂为双季戊四醇、三羟甲基丙烷或者季戊四醇中的一种。
13.步骤(2)中的助剂包括热稳定剂、抗氧化剂;热稳定剂的用量为低聚酯化物a和低聚酯化物b总质量的0.01%-0.1%,抗氧化剂的用量为低聚酯化物a和低聚酯化物b总质量的0.01%-0.1%。
14.抗氧化剂优选为2,6-二叔丁基-4-甲基苯酚、三(2,4-二叔丁基苯基)亚磷酸酯、四[β-(3,5-二叔丁基-4-羟基苯基)丙酸]季戊四醇酯中的一种。
[0015]
热稳定剂优选为磷酸三甲酯、磷酸三苯酯和磷酸乙酯中的至少一种。
[0016]
所述的端羧基有机硅氧烷的制备方法为:将四甲基二硅氧烷、甲基丙烯酸、催化剂b混合,升温至60-65℃反应1-2h后,继续升温至85-95℃反应2-3h,最后降至40℃以下,经过减压蒸馏,得到端羧基有机硅氧烷;化学反应方程式如下:
[0017]
。
[0018]
四甲基二硅氧烷与甲基丙烯酸的摩尔比为1:(1.1-2.5);催化剂b以铂来计,是四甲基二硅氧烷和甲基丙烯酸总质量的2ppm-4ppm;步骤(1)中低聚酯化物a和低聚酯化物b的反应条件均为:加热至180-200℃,酯化脱水反应1-2h,继续升温至250-270℃反应2-3h,反应压力为0.1~0.25mpa。
[0019]
步骤(2)中缩聚反应的温度为275-285℃,反应时间为180-200min,真空度为-0.098~-0.1mpa。
[0020]
步骤(1)中低聚酯化物a中对苯二甲酸、端羧基有机硅氧烷的重量比为1:(0.01-0.2);对苯二甲酸、新戊二醇、乙二醇的摩尔比为1:(1.1-2.0):(0.3-0.6)。
[0021]
步骤(1)中低聚酯化物b中对苯二甲酸、端羧基有机硅氧烷的重量比为1:(0.01-0.2);对苯二甲酸与乙二醇的摩尔比为1:(1.1-2.0)。
[0022]
步骤(1)中新戊二醇的摩尔量为低聚酯化物a和低聚酯化物b的所有新戊二醇和乙二醇总摩尔量的10-40%。
[0023]
步骤(1)中制备低聚酯化物a的催化剂a的加入量为低聚酯化物a质量的0.03-0.3%;步骤(1)制备低聚酯化物b的催化剂a的加入量为低聚酯化物b质量的0.03-0.3%;步骤(2)中催化剂a的加入量为低聚酯化物a和低聚酯化物b总质量的0.03-0.3%。
[0024]
步骤(1)中制备低聚酯化物a和低聚酯化物b的交联剂加入量为各自加入原料对苯二甲酸摩尔量的0.1-0.8%。
[0025]
所述的改性petg共聚酯是上述的制备方法制得的。
[0026]
采用所述的改性petg共聚酯的发泡颗粒的制备方法:将改性petg共聚酯放入70-90℃的烘箱烘干4-6h,再将其与物理发泡剂超临界状态的co2放入高压釜,升温至70-90℃,排除反应釜内的空气,然后密封反应釜,保持压力为10-20mpa,恒温恒压发泡4-6h,得到改性petg共聚酯的发泡颗粒。
[0027]
具体的,所述的改性petg共聚酯发泡颗粒的制备方法,包括以下步骤:1)制备端羧基有机硅氧烷:将四甲基二硅氧烷、甲基丙烯酸、催化剂b混合,升温至60-65℃反应1-2h后,继续升温至85-95℃反应2-3h,进行硅氢加成反应,反应完毕,降至40℃以下,再升温至120℃,减压蒸馏,得到端羧基有机硅氧烷;2)分酯化反应:将对苯二甲酸、端羧基有机硅氧烷、新戊二醇、乙二醇、催化剂a混合升温至195-260℃,反应压力0.1-0.25mpa,进行酯化反应180-260min,得到低聚酯化物a;将对苯二甲酸、端羧基有机硅氧烷、乙二醇混合,与上述相同的反应条件进行酯化反应,得到低聚酯化物b。
[0028]
3)缩聚反应:将低聚酯化物a和低聚酯化物b混合,加入催化剂a、助剂,升温至275-285℃,保持真空度为-0.098~-0.1mpa,进行缩聚反应180-200min,得到改性petg共聚酯。
[0029]
4)发泡:将改性petg共聚酯放入70~90℃的烘箱烘干4-6h,再将其与物理发泡剂超临界状态的co2放入高压釜,排除反应釜内的空气,然后密封反应釜,将反应釜升温至70-90℃,保持压力为10-20mpa,恒温恒压发泡4-6h,得到改性petg共聚酯发泡颗粒。
[0030]
与现有技术相比,本发明具有的有益效果是:(1)本发明在工艺上不同于传统的“一步法”,采用了分酯化工艺,将新戊二醇与乙二醇分开反应,使得petg产品中npg的含量稳定,不会出现因npg与乙二醇竞聚的问题出现的npg含量波动问题。
[0031]
(2)本发明通过共聚引入有机硅氧烷基团,提升了petg材料对co2的亲和性,使得聚合物中co2的溶解度提高,因而更有利于气泡成核,使得petg材料能进行超临界co2发泡。发泡后的材料发泡倍率高,泡孔直径小,并且发泡更加均匀,从而提升了材料的冲击强度。并且相比之前的挤出发泡在工艺上更为简便,避免了二次加工。
[0032]
(3)本发明制备的改性petg共聚酯,引入含硅烷结构的柔顺链段使得petg在低温下的耐冲击性能更为优异。
具体实施方式
[0033]
为了便于理解本发明,本发明列举实施例如下。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。所用原料均来自市售产品。
[0034]
实施例1所述的改性petg共聚酯发泡颗粒的制备方法,包括以下步骤:
1)制备端羧基有机硅氧烷:将四甲基二硅氧烷134.33g、甲基丙烯酸94.70g、铂(0)-1,3-二乙烯基四甲基二硅氧烷络合物(含pt量19.0-21.5%)0.0025g混合,升温至60℃反应1h后,继续升温至85℃反应2h,进行硅氢加成反应,反应完毕,降至40℃以下,再升温至120℃,减压蒸馏,得到端羧基有机硅氧烷。
[0035]
2)分酯化反应:将166.2g对苯二甲酸、1.67g端羧基有机硅氧烷、114.57g新戊二醇、18.62g乙二醇、0.08g钛酸四丁酯、0.14g三羟甲基丙烷混合升温至180℃,脱水反应1h;继续升温至250℃反应2h,反应压力0.1mpa,得到低聚酯化物a;将1495.18g对苯二甲酸、134.57g端羧基有机硅氧烷、614.48g乙二醇、0.57g钛酸四丁酯、1.21g三羟甲基丙烷混合,与上述相同的反应条件进行酯化反应,得到低聚酯化物b。
[0036]
3)缩聚反应:将低聚酯化物a和低聚酯化物b混合,加入0.65g钛酸四丁酯、0.22g磷酸三苯酯、0.22g2,6-二叔丁基-4-甲基苯酚,升温至275℃,保持真空度为-0.098mpa,进行缩聚反应180min,得到改性petg共聚酯。
[0037]
4)发泡:将1000g改性petg共聚酯放入70℃的烘箱烘干4h,再将其与400g物理发泡剂超临界状态的co2放入高压釜,排除反应釜内的空气,然后密封反应釜,将反应釜升温至70℃,保持压力为10mpa,恒温恒压发泡4h,得到改性petg共聚酯发泡颗粒。
[0038]
实施例2所述的改性petg共聚酯发泡颗粒的制备方法,包括以下步骤:1)制备端羧基有机硅氧烷:将四甲基二硅氧烷268.67g、甲基丙烯酸344.36g、铂、氯铂酸的异丙醇溶液0.007g混合,升温至63℃反应1.5h后,继续升温至90℃反应2.5h,进行硅氢加成反应,反应完毕,降至40℃以下,再升温至120℃,减压蒸馏,得到端羧基有机硅氧烷。
[0039]
2)酯化反应:将332.27g对苯二甲酸、33.23g端羧基有机硅氧烷、312.46g新戊二醇、31.04g乙二醇、0.19g醋酸锑、0.27g双季戊四醇混合升温至190℃,脱水反应1.5h;继续升温至260℃反应2.5h,反应压力0.1mpa,得到低聚酯化物a;将1329.05g对苯二甲酸、132.91g端羧基有机硅氧烷、744.82g乙二醇、0.58g醋酸锑、1.08g双季戊四醇混合,与上述相同的反应条件进行酯化反应,得到低聚酯化物b。
[0040]
3)缩聚反应:将低聚酯化物a和低聚酯化物b混合,加入0.76g醋酸锑、0.26g磷酸三甲酯、0.26g三(2,4-二叔丁基苯基)亚磷酸酯,升温至280℃,保持真空度为-0.099mpa,进行缩聚反应190min,得到改性petg共聚酯。
[0041]
4)发泡:将1000g改性petg共聚酯放入80℃的烘箱烘干5h,再将其与400g物理发泡剂超临界状态的co2放入高压釜,排除反应釜内的空气,然后密封反应釜,将反应釜升温至80℃,保持压力为15mpa,恒温恒压发泡5h,得到改性petg共聚酯发泡颗粒。
[0042]
实施例3所述的改性petg共聚酯发泡颗粒的制备方法,包括以下步骤:1)制备端羧基有机硅氧烷:将四甲基二硅氧烷268.66g、甲基丙烯酸430.45g、铂(0)-1,3-二乙烯基四甲基二硅氧烷络合物(含pt量19.0-21.5%)0.015g混合,升温至65℃反应2h后,继续升温至95℃反应3h,进行硅氢加成反应,反应完毕,降至40℃以下,再升温至120℃,减压蒸馏,得到端羧基有机硅氧烷。
[0043]
2)酯化反应:将664.53g对苯二甲酸、132.91g端羧基有机硅氧烷、833.2g新戊二
醇、37.25乙二醇、0.45g乙二醇锑、0.54g季戊四醇混合升温至200℃,脱水反应2h;继续升温至270℃反应3h,反应压力0.15mpa,得到低聚酯化物a;将996.79g对苯二甲酸、199.36g端羧基有机硅氧烷、744.82g乙二醇、0.52g乙二醇锑、0.81g季戊四醇混合,与上述相同的反应条件进行酯化反应,得到低聚酯化物b。
[0044]
3)缩聚反应:将低聚酯化物a和低聚酯化物b混合,加入0.97g乙二醇锑、0.36g磷酸乙酯、0.36g四[β-(3,5-二叔丁基-4-羟基苯基)丙酸]季戊四醇酯,升温至285℃,保持真空度为-0.1mpa,进行缩聚反应200min,得到改性petg共聚酯。
[0045]
4)发泡:将1000g改性petg共聚酯放入90℃的烘箱烘干6h,再将其与400g物理发泡剂超临界状态的co2放入高压釜,排除反应釜内的空气,然后密封反应釜,将反应釜升温至90℃,保持压力为20mpa,恒温恒压发泡6h,得到改性petg共聚酯发泡颗粒。
[0046]
实施例4所述的改性petg共聚酯发泡颗粒的制备方法,包括以下步骤:1)制备端羧基有机硅氧烷:将四甲基二硅氧烷268.66g、甲基丙烯酸344.36g、铂(0)-1,3-二乙烯基四甲基二硅氧烷络合物(含pt量19.0-21.5%)0.01g混合,升温至65℃反应2h后,继续升温至90℃反应3h,进行硅氢加成反应,反应完毕,降至40℃以下,再升温至120℃,减压蒸馏,得到端羧基有机硅氧烷。
[0047]
2)酯化反应:将332.27g对苯二甲酸、33.23g端羧基有机硅氧烷、312.45g新戊二醇、31.04g乙二醇、0.61g硅酸四乙酯、1.35g三羟甲基丙烷混合升温至190℃,脱水反应1.5h;继续升温至260℃反应2.5h,反应压力0.1mpa,得到低聚酯化物a;将1329.05g对苯二甲酸、132.91g端羧基有机硅氧烷、744.82g乙二醇、1.92g硅酸四乙酯、5.37g三羟甲基丙烷混合,与上述相同的反应条件进行酯化反应,得到低聚酯化物b。
[0048]
3)缩聚反应:将低聚酯化物a和低聚酯化物b混合,加入2.53g硅酸四乙酯、1.27g磷酸三苯酯、1.27g2,6-二叔丁基-4-甲基苯酚,升温至280℃,保持真空度为-0.1mpa,进行缩聚反应190min,得到改性petg共聚酯。
[0049]
4)发泡:将1000g改性petg共聚酯放入80℃的烘箱烘干5h,再将其与400g物理发泡剂超临界状态的co2放入高压釜,排除反应釜内的空气,然后密封反应釜,将反应釜升温至80℃,保持压力为15mpa,恒温恒压发泡5h,得到改性petg共聚酯发泡颗粒。
[0050]
实施例5所述的改性petg共聚酯发泡颗粒的制备方法,包括以下步骤:1)制备端羧基有机硅氧烷:将四甲基二硅氧烷268.66g、甲基丙烯酸344.36g、铂(0)-1,3-二乙烯基四甲基二硅氧烷络合物(含pt量19.0-21.5%)0.01g混合,升温至65℃反应2h后,继续升温至90℃反应3h,进行硅氢加成反应,反应完毕,降至40℃以下,再升温至120℃,减压蒸馏,得到端羧基有机硅氧烷。
[0051]
2)酯化反应:将332.27g对苯二甲酸、33.23g端羧基有机硅氧烷、312.45g新戊二醇、31.04g乙二醇、1.82g钛酸四丁酯、2.15g三羟甲基丙烷混合升温至190℃,脱水反应1.5h;继续升温至260℃反应2.5h,反应压力0.1mpa,得到低聚酯化物a;将1329.05g对苯二甲酸、132.91g端羧基有机硅氧烷、744.82g乙二醇、5.76g钛酸四丁酯、1.08g三羟甲基丙烷混合,与上述相同的反应条件进行酯化反应,得到低聚酯化物b。
[0052]
3)缩聚反应:将低聚酯化物a和低聚酯化物b混合,加入7.58g钛酸四丁酯、2.53g磷
酸三苯酯、2.53g2,6-二叔丁基-4-甲基苯酚,升温至280℃,保持真空度为-0.1mpa,进行缩聚反应190min,得到改性petg共聚酯。
[0053]
4)发泡:将1000g改性petg共聚酯放入80℃的烘箱烘干5h,再将其与400g物理发泡剂超临界状态的co2放入高压釜,排除反应釜内的空气,然后密封反应釜,将反应釜升温至80℃,保持压力为15mpa,恒温恒压发泡5h,得到改性petg共聚酯发泡颗粒。
[0054]
对比例1一种petg共聚酯的制备方法与实施例1酯化反应和缩聚反应相同,但是将端羧基有机硅氧烷去掉,制得一种petg共聚酯;一种petg共聚酯发泡颗粒的制备方法,是采用上述制备的petg共聚酯,与实施例1相同的发泡步骤,制得一种petg共聚酯发泡颗粒。
[0055]
对比例2一种petg共聚酯的制备方法与实施例1酯化反应和缩聚反应相同,但是将端羧基有机硅氧烷去掉,制得一种petg共聚酯;一种petg共聚酯发泡颗粒的制备方法:采用上述制备的petg共聚酯,采用连续挤出发泡工艺,加入1000g改性petg共聚酯,10g双(3,4-环氧环己基甲基)己二酸酯作为扩链剂,设定温度为220℃,螺杆转速为50r/min,挤出时间20min,制得一种petg共聚酯发泡颗粒。
[0056]
对比例3一种petg共聚酯的制备方法与实施例1酯化反应和缩聚反应相同,但是将端羧基有机硅氧烷去掉,制得一种petg共聚酯;一种petg共聚酯发泡颗粒的制备方法:采用上述制备的petg共聚酯,采用挤出发泡工艺,加入1000g改性petg共聚酯,温度设为220℃,螺杆转速为50r/min,挤出时间20min ,制得一种petg共聚酯发泡颗粒。
[0057]
对比例4一种petg共聚酯的“一步法”制备方法,包括以下步骤:(1)酯化反应:将1661.31g对苯二甲酸(pta)、16.62g端羧基有机硅氧烷、104.15g新戊二醇(npg)、558.62g乙二醇(eg)、0.60g钛酸四丁酯和1.34g三羟甲基丙烷混合,升温至180℃,脱水反应1h;继续升温至250℃反应2h,反应压力0.1mpa,得到酯化物。
[0058]
(2)缩聚反应:将步骤(1)中酯化物与0.60g钛酸四丁酯、0.20g抗氧化剂、0.20g热稳定剂混合,加热至275℃,反应时间为180min,真空度控制在-0.098mpa,得到petg共聚酯。
[0059]
本对比例的petg共聚酯发泡颗粒的制备方法与实施例1完全相同。
[0060]
将以上实施例1-5与对比例1-4制备的改性petg共聚酯的npg含量、冲击强度性能,以及发泡颗粒的发泡倍率、co2溶解度进行性能测试,其测试结果如下表2所示。
[0061]
首先对制备的共聚酯进行预处理,然后再检测,粒料预处理:在成型试样前,粒料在真空干燥箱内进行预热干燥处理,装盘厚度小于4cm,在80℃下连续干燥5h。
[0062]
1、npg含量测试重复三次实施例1及对比例4的实验方法后分别测试,按照gb/t 14190-2017标准中deg的测试方法,在岛津lc-14b气相色谱上进行测试。以1,6-己二醇(色谱纯)为内标液,气相色谱担体为20~40目白色硅藻土,气相色谱固定液为peg-20m(分子质量20000)。
[0063]
气相色谱仪测试条件:氮气进气速率25~35ml/min,氢气进气速率20~30ml/min,空气进气速率700~900ml/min,温柱165~170℃,检测器温度175~185℃,汽化室温度200~210℃,注入量1~2μl,色谱柱为直径3.5mm、长2250mm的装有浓度为5%的peg-20m和白色硅藻土担体(20~40目)的玻璃柱。
[0064]
新戊二醇在petg中质量含量为:
[0065]
式中,x0—样品的新戊二醇质量含量;a1—新戊二醇峰面积,单位为平方厘米(cm2);a
st
—1,6-己二醇峰面积,单位为平方厘米(cm2);m—试样的质量,单位为克(g);k—工作曲线的斜率,单位为毫克(mg);k是以校正溶液中的新戊二醇的质量(单位为毫克)为纵坐标,新戊二醇峰面积与1,6-己二醇峰面积之比为横坐标,线性回归绘制校正曲线,且相关系统高于0.99的工作曲线的斜率。
[0066]
2、冲击强度注塑试样按gb/t17037.1-1997规定进行,用gb/t17037.1-1997中的b型模具制备符合gb/t1040.2-2006中b1型试样。试样应按iso 2818:1994由板材机加工而成,尽可能使用a型缺口。测试时,无缺口试样的机加工表面不应处于拉伸状态。
[0067]
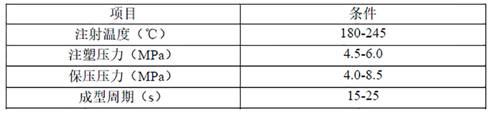
试样制备条件:采用fanuc roboshot ɑ-s100ia日本发那科全电动注塑机,螺杆注射机工艺,工艺参数如表1所示:表1 注塑机工艺参数
[0068]
试样的状态调节和试验的标准环境:试样的状态调节按gb/t2918-1998的规定进行,状态调节的条件为温度23℃
±
2℃,调节时间48h,试验在gb/t2918-1998规定的标准环境下进行,环境的温度为23℃
±
2℃,相对湿度为50%
±
10%。
[0069]
3、发泡倍率利用排水法得到petg基础树脂以及petg发泡珠粒的密度,按照gb/t6343-2009中介绍方法测量。发泡倍率计算:b=ρ1/ρ2,其中,b为发泡倍率,ρ1为petg基础树脂的密度,ρ2为发泡材料的密度。
[0070]
4、co2溶解度按照gb/t 32198-2015标准中用衰减全反射法(atr)进行定量分析,将压膜机温度
设定在95℃,压力设定为6mpa,使用开孔的金属垫片压制厚度为50μmpetg聚合物薄膜,保证红外光谱仪在氮气吹扫环境下进行测试。
[0071]
表2 petg共聚酯及其发泡颗粒的性能测试结果
[0072]
由表2可以看出,比较实施例1与对比例1可知,加入端羧基有机硅氧烷,可有效提高材料对co2的溶解度。实施例1-5与对比例1分别采用了不同量的端羧基有机硅氧烷,比较可知,聚合时使用端羧基有机硅氧烷的聚合产物冲击强度并未降低,反而出现较大幅度提升,在低温环境下表现依然优异。实施例1与对比例2、3对比可知,超临界co2发泡工艺获得的发泡材料发泡倍率明显高于传统挤出发泡,泡孔直径小,没有破裂。实施例1与对比例4比较可知,利用分酯化制得的petg中npg的含量要更加稳定。
[0073]
当然,上述内容仅为本发明的较佳实施例,不能被认为用于限定对本发明的实施例范围。本发明也并不仅限于上述举例,本技术领域的普通技术人员在本发明的实质范围内所做出的均等变化与改进等,均应归属于本发明的专利涵盖范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1