一种抗包虫病药物及其制备方法和医药用途
本发明涉及药物化学领域,具体涉及一系列β-咔啉衍生物及其制备方法和医药用途,对β-咔啉不同位点进行修饰,设计、合成了一系列β-咔啉类似物,以及这些化合物的医药用途,特别是在制备预防或治疗包虫病的药物中的应用。
背景技术:
1、包虫病又称棘球蚴病,棘球蚴病因宿主感染棘球绦虫成虫或续绦期幼虫所致,为涉及众多宿主的人兽共患病(lancet,2003,362:1295-304)。在我国主要有两型:囊型包虫病(cystic echinococcosis,ce)由细粒棘球蚴绦虫(echinococcus granulosus,e.g)引起;泡型包虫病(alveolar echinococcosis,ae)由多房棘球蚴引起。该病好发于畜牧业发达地区,是全球性的公共卫生问题(int.j.infec.des.2019,79,89)。在我国,囊型包虫病主要分布于西北和西南地区,其中青藏高原地区为该病的重流行区(adv.parasitol.2017,95,315-493)。迄今为止,手术治疗是包虫病的首选治疗方法,但是该方法存在一定风险,并且对医疗条件和医生技术有较高要求(acta trop.2020,203:105283)。因此,药物治疗作为一种辅助治疗手段越来越引起人们的重视。
2、骆驼蓬(peganum harmala l.)是蒺藜科、骆驼蓬属多年生草本植物,主要分布于我国华北及西北地区(food ch&em.toxicol.2010,48(3),839-845;food ch&em.toxicol.2017,103,261-269;toxins(basel).2015,7(11),4507-4518)。β-咔啉是骆驼蓬科植物骆驼莲的种子中一种三元环生物碱,为苯并吲哚类结构。它能抑制拓扑异构酶、细胞周期蛋白依赖性激酶和dna的合成,并能嵌入dna中,因此具有广泛的生物学活性。研究表明,β-咔啉衍生物去氢骆驼蓬碱(harmine,hm)具抗寄生虫(tetrah&edron lett.2010,51(4),583-585;j.drug target.2004,12(3),165-175)、杀菌(fitoterapia.2010,81(7),779-782)、抗肿瘤(oncol.rep.2017,38(5),2927-2934;phytomedicine.2017,28,10-18;altern.med.2017,1-7)、抗抑郁(prog.neuropsychopharmacol.biol.psychiatry.2017,79(pt b),258-267;brain res.bull.2018,137,294-300)等多种药理作用。课题组发现,hm具有较好的抗包虫病活性,并且去氢骆驼蓬碱和临床上常用的抗包虫病药物阿苯达唑(albendazole,abz)联合用药能够发挥协同作用。但hm具有一定的神经毒性,会引起患者呕吐、震颤、幻觉甚至死亡,因此限制了临床应用和发展。
3、
4、基于此,课题组围绕hm母核,对β-咔啉环1、2、3、7、9等五个结构位点进行结构修饰改造,旨在提高治疗囊型包虫病活性同时降低其神经毒性(专利号:cn105998014b)。团队前期研究发现hm衍生物dh-330和dh-004对ce有较好的疗效,它们的lc50值分别41.55±9.48μm、47.77±18.99μm,优于hm(lc50=250.39±92.11μm,专利号:cn105998014a)。但是在25mg/kg低剂量下,dh-330对小鼠细粒棘球蚴生长抑制率仅有32.91%,众所周知,包虫病的治疗是一个长期用药的过程,因此如何发现更有效的衍生物,从而进一步降低剂量或减少服药疗程,提高患者依从性和减少不良反应的发生还需要更深入研究。
技术实现思路
1、根据以上背景,本发明对β-咔啉不同位点进行修饰,设计并合成了一类对细粒棘球蚴具有杀伤作用的抗包虫病化合物,并提供了上述化合物的制备方法,以及含这些化合物的药用组合物、其药学上可接受的盐,及其医药用途。
2、技术方案:
3、第一方面,本发明公开了一种化合物,为式ⅰ、ⅱ所示的化合物,或其药学上可接受的盐:
4、
5、其中,r1选自或-ch3;
6、r2选自
7、r3选自
8、r4选自
9、在一些实施例中,式ⅰ所示的化合物选自:1a:4-((4-羟基哌啶-1-基)甲基)-n-(1-(4-甲氧基苯基)-9h-吡啶并[3,4-b]吲哚-3-基)苯甲酰胺
10、
11、1b:n-(1-(3-甲氧基苯基)-9h吡啶[3,4-b]吲哚-3-基)-4-((4-甲基-1,4-二氮杂-1-基)甲基)苯甲酰胺
12、
13、1c:4-([1,4'-联哌啶]-1'-基甲基)-n-(1-(3,4-二甲氧基苯基)-9h吡啶[3,4-b]吲哚-3-基)苯甲酰胺
14、
15、1d:n-(1-(3,4-二甲氧基苯基)-9h吡啶并[3,4-b]吲哚-3-基)-4-((4-甲基-1,4-二氮杂-1-基)甲基)苯甲酰胺
16、
17、1e:4-([1,4'-联哌啶]-1'-基甲基)-n-(1-甲基-9h-吡啶[3,4-b]吲哚-3-基)苯甲酰胺
18、
19、1f:4-([1,4'-联哌啶]-1'-基甲基)-n-(1-(对甲苯基)-9h吡啶[3,4-b]吲哚-3-基)苯甲酰胺
20、
21、1g:4-((4-甲基-1,4-二氮杂-1-基)甲基)-n-(1-(3,4,5-三甲氧基苯基)-9h吡啶[3,4-b]吲哚-3-基)苯甲酰胺
22、
23、在一些实施例中,式ⅱ所示的化合物选自:2a:n-(4-([1,4'-联哌啶]-1'-基甲基)苯基)-1-(4-甲氧基苯基)-9h吡啶[3,4-b]吲哚-3-甲酰胺
24、
25、2b:n-(4-([1,4'-联哌啶]-1'-基甲基)苯基)-1-(3-甲氧基苯基)-9h吡啶[3,4-b]吲哚-3-甲酰胺
26、
27、2c:n-(4-([1,4'-联哌啶]-1'-基甲基)苯基)-1-(3,4-二甲氧基苯基)-9h-吡啶并[3,4-b]吲哚-3-甲酰胺
28、
29、2d:n-(4-([1,4'-联哌啶]-1'-基甲基)苯基)-1-(2-甲氧基苯基)-9h吡啶[3,4-b]吲哚-3-甲酰胺
30、
31、2e:n-(4-([1,4'-联哌啶]-1'-基甲基)苯基)-1-(2,3-二甲氧基苯基)-9h-吡啶并[3,4-b]吲哚-3-甲酰胺
32、
33、2f:n-(4-([1,4'-联哌啶]-1'-基甲基)苯基)-1-(2,4-二甲氧基苯基)-9h吡啶[3,4-b]吲哚-3-甲酰胺
34、
35、2g:n-(4-([1,4'-联哌啶]-1'-基甲基)苯基)-1-(3,5-二甲氧基苯基)-9h-吡啶并[3,4-b]吲哚-3-甲酰胺
36、
37、第二方面,本发明还提供了所述化合物的制备方法。
38、化合物ⅰ的制备方法包括:
39、化合物1-1r1cho和l-色氨酸在酸性条件下进行pictet-spengler反应得中间体1-2;
40、中间体1-2的羧基转化为甲酯得中间体1-3;
41、中间体1-3氧化得到中间体1-4;中间体1-4经水合肼处理后转化为中间体1-5,中间体1-5的肼基在nano2存在条件下转化为叠氮基生成中间体1-6,中间体1-6通过curtis重排转化为氨基生成中间体1-7;中间体1-7和对氯甲基苯甲酰氯反应生成中间体1-8;中间体1-8与化合物1-9r2h发生亲核取代反应得目标化合物ⅰ;
42、
43、在一些实施例中,式ⅰ所示化合物可用如下方法制备得到:以l-色氨酸为原料与化合物1-1(相应的醛r1cho)进行pictet-spengler反应生成中间体1-2,然后将其转化为甲酯中间体1-3。中间体1-3在dmf中被kmno4氧化得到中间体1-4,经水合肼处理后转化为中间体1-5。1-5的肼基在nano2存在时转化为叠氮基生成1-6,在h2o中hac存在时通过curtis重排转化为氨基生成中间体1-7。在dcm中1-7和对氯甲基苯甲酰氯在tea存在下反应生成中间体1-8。在k2co3和ki存在下,中间体1-8与化合物1-9(不同仲胺r2h)在乙腈中发生亲核取代反应,形成目标化合物ⅰ。
44、在一些实施例中,化合物ⅰ的合成路线包括:
45、
46、化合物ⅱ的制备方法包括:
47、化合物2-1r3cho和l-色氨酸在酸性条件下进行pictet-spengler反应得中间体2-2,
48、中间体2-2氧化得到中间体2-3;
49、4-硝基苄溴与化合物2-4r4h发生亲核取代反应得到中间体2-5,中间体2-5的硝基还原为氨基得到中间体2-6;
50、中间体2-3和中间体2-6通过酰胺缩合反应生成目标化合物ⅱ;
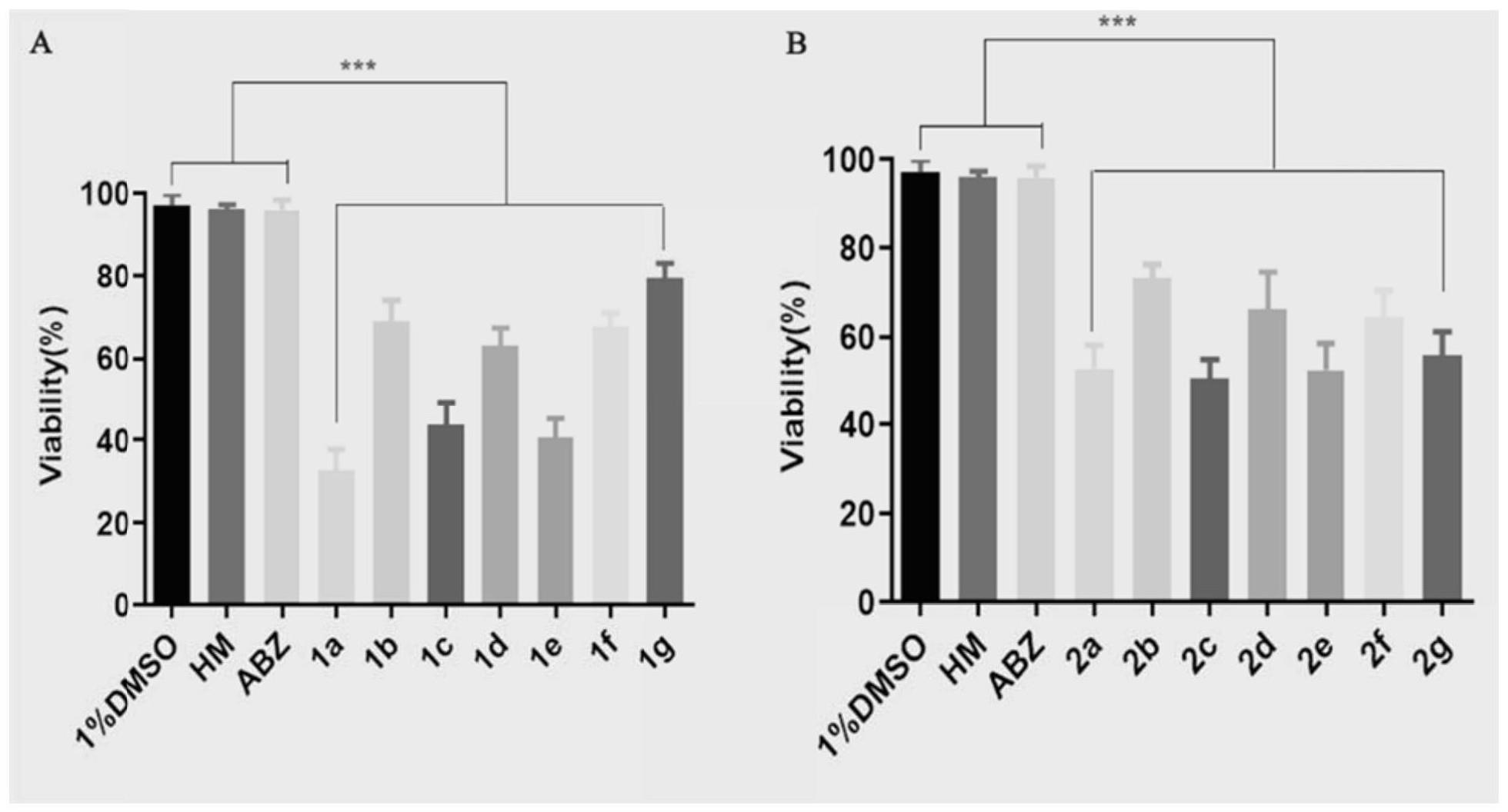
51、
52、在一些实施例中,式ⅱ所示化合物可用如下方法制备得到:以l-色氨酸为原料与化合物2-1(相应的醛r3cho)进行pictet-spengler反应生成2-2。中间体2-2在dmf被kmno4氧化得到中间体2-3。在k2co3和ki存在下,4-硝基苄溴在乙腈中与化合物2-4r4h仲胺发生亲核取代反应得到中间体2-5,然后在ch3oh中与pd/c和h2还原得到中间体2-6。在edci和dmap存在的情况下,中间体2-3与dcm中的中间体2-6通过酰胺缩合反应生成目标化合物ⅱ。
53、在一些实施例中,化合物ⅱ的合成路线包括:
54、
55、第三方面,本发明还提供一种药物组合物,包括含有治疗有效量的所述化合物或其药学上可接受的盐,以及药学上可接受的载体或辅料。
56、本发明药物组合物的剂型可以由本领域技术人员,按照药学领域的常规方法制备。例如,使活性成分与一种或多种载体(也称为辅料)混合,然后将其制成所需的剂型,包括片剂、胶囊、颗粒剂、气雾剂;还可以按照注射剂常规生产方法制成静脉注射或静脉注射冻干剂。
57、第四方面,本发明还提供所述化合物、药物组合物在制备预防或治疗包虫病的药物中的应用。
58、有益效果:本发明的化合物具有如下优异的性能:(1)化合物稳定性较高。(2)化合物在在合成上具有易可获得性。(3)化合物1a、1c、1e可有效抑制细粒棘球蚴生长。(4)化合物1a抑制细粒棘球蚴的生长具有浓度依赖性,低浓度下也能完全抑制细粒棘球蚴生长。(5)化合物1a能抑制细粒棘球蚴生长,且优于阳性药阿苯达唑(abz)以及化合物dh-003。(6)与去氢骆驼蓬碱相比,化合物1a具有良好的安全性。
59、经过实验验证,本发明的化合物也具有如下优异的性能:1a被证明是最佳化合物,初始浓度为1μm时,发现其中衍生物1a干预2天后有超过50%的细粒棘球蚴原节头(pscs)死亡,并且具有剂量依赖性。此外,与abz和hm对照组相比,1a还表现出明显的形态学改变。最后在衍生物干预细粒棘球蚴原节头实验中,1a可能导致原节头损伤和活力丧失。本次实验中,β-咔啉衍生物1a的体外活性优于阳性药阿苯达唑(abz)和先导化合物去氢骆驼蓬碱(hm)。随后,对1a进行了体内药效学研究。结果表明1a处理的小鼠与模型对照组相比,囊重减少量具有统计学意义,且处理28天后与abz和hm处理组相比也具有统计学意义。处理14天,1a低剂量(12.5mg/kg)囊湿重减少率与abz和hm高剂量组(50mg/kg)相当;处理28天,1a囊湿重减少率最高可达76.87%,分别是同剂量下abz和hm处理组的1.39倍、1.35倍。因此,1a具有非常显著的抗包虫效果。总的来说,我们的研究表明1a是对抗包虫病药物中非常有希望的候选药物。
- 还没有人留言评论。精彩留言会获得点赞!