一种结合二代和三代测序的多菌株全基因组分析方法
本发明属于环境微生物领域,公开了一种结合二代和三代测序的多菌株全基因组分析方法。
背景技术:
1、微生物菌株全基因组测序能够全面、精确分析细菌基因组的碱基组成,是掌握菌株系统分类、基因突变、代谢功能等的重要手段。菌株全基因组测序分析方法目前流行的有3种,即二代测序、三代测序以及传统二代-三代混合测序。二代测序技术成本较低,但由于读长短,生成的基因组数据完整性和拼装质量较差,组装获得的基因组通常存在较多的缺失序列。因此,二代测序很难或者几乎不能获得完整的环状dna,对菌株基因组的测序完整度一般只能达到90%左右。为填补上述缺失序列、获得完成的基因组,还需要进行一代测序或三代测序。其中,一代测序填补缺失序列的方法非常繁琐,涉及特异性引物设计、克隆文库建立、蓝白斑筛选、一代测序、序列拼装等复杂过程。一代测序填补缺失序列需要在每个缺失序列前后设计引物,建立克隆文库,最后通过一代测序获得缺失序列的序列信息。三代测序技术具有长读长的优势,但价格比较昂贵,常用的技术包括pacbio和oxfordnanopore。其中,oxford nanopore技术可以直接对dna进行测序,样品输入量要求更低,设备便携,测序速度快,价格相对于pacbio技术较低,更适合微生物的基因组研究。除了价格昂贵,三代测序还存在碱基错误率高的问题。传统二代-三代混合测序技术的测序质量和基因组完整度都较高。
2、在环境微生物的纯菌研究中,经常需要对多个菌株的全基因组进行分析,需要同时大批量获得多个菌株的完成的、封闭成环的基因组完成图。针对上述情况,传统的二代-三代混合测序方法用于环境微生物测序的费用非常昂贵。因此,亟需一种针对环境微生物更廉价的测序分析方法。
技术实现思路
1、本发明的目的在于解决现有技术中存在的问题,提供了一种结合二代和三代测序的多菌株全基因组分析方法。
2、为了实现上述发明目的,本发明所用的具体技术方案如下:
3、本发明提供一种结合二代和三代测序的多菌株全基因组分析方法,步骤包括:
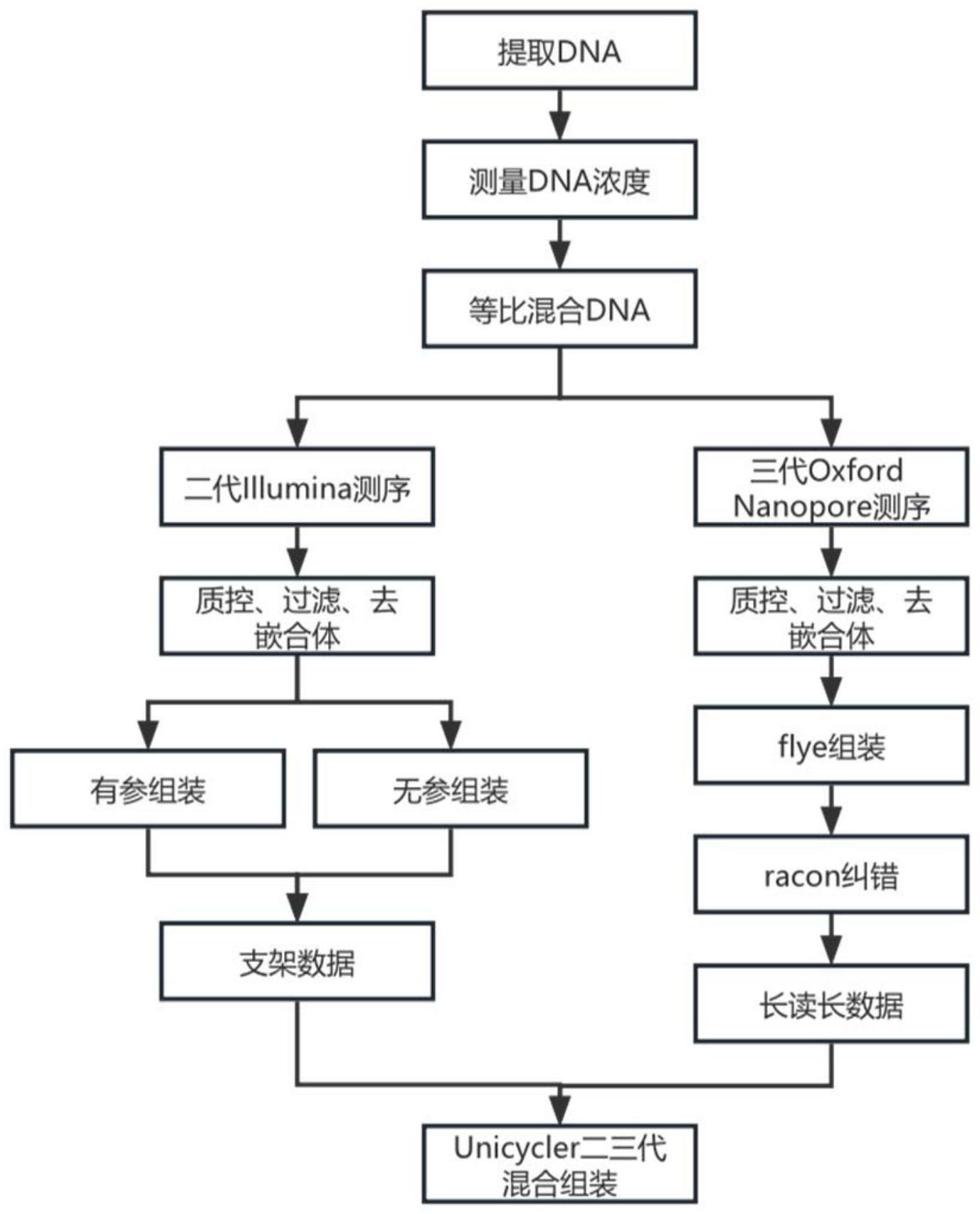
4、s1:提取多个菌株的基因组dna,得到每个菌株的单独dna样品,并测定单独dna样品中的dna浓度。将多个菌株的单独dna样品进行二代测序得到单独样品下机数据,对单独样品下机数据分别进行质量控制、过滤和去嵌合体后,得到单独样品读长。
5、对于有参考基因组的菌株,采用sibelia算法进行组装。对于无参考基因组的菌株,采用de bruijn图算法进行组装。最终获得每个菌株的支架数据,即获得基因组草图。
6、s2:将单独dna样品根据dna浓度等质量比混合,获得多个菌株的dna混合样品。将多个菌株的dna混合样品进行三代测序得到混合样品下机数据,对混合样品下机数据进行质量控制、过滤和去嵌合体后,得到混合样品有效数据。对混合样品有效数据进行修剪和组装,获得三代测序组装dna混合样品的长读长。
7、s3:利用三代测序获得的长读长数据,逐一填补步骤s1中二代测序获得的每个菌株支架数据中的缺失序列,从而获得完整、封闭成环的菌株基因组完整图。
8、作为优选,上述二代测序采用illumina二代测序,三代测序采用oxford nanopore三代测序。
9、作为优选,上述s1中的dna浓度采用生物检测分析仪检测。
10、作为优选,上述每个菌株的illumina二代测序数据量为6.0gb。
11、作为优选,上述illumina二代测序得到的单独样品下机数据使用fastqc软件进行首次质量控制,使用fastp软件进行过滤,再次使用fastqc软件进行二次质量控制,得到illumina二代测序读长序列。使用metaspades软件,将illumina二代测序获得的读长序列组装成菌株支架数据。
12、作为优选,上述oxford nanopore三代测序数据量范围为(0.05~0.1)×mgb,其中m为dna混合样品中待分析的菌株数量。
13、作为优选,上述oxford nanopore三代测序得到的混合样本下机数据使用nanoplot软件进行首次质量控制,使用filtlong软件进行数据过滤,再次使用nanoplot软件进行二次质量控制。
14、作为优选,上述s2中的混合样品有效数据进行修剪和组装的过程采用flye软件。
15、作为优选,上述三代测序组装dna混合样品长读长序列的过程使用flye软件。
16、作为优选,上述填补过程使用unicycler软件,利用oxford nanopore三代测序产生的长读长序列,对illumina二代测序生成的每个菌株的支架数据逐一进行拼装和缺失序列填补,提高基因组的组装质量和连续性。在填补过程中,长读长序列一旦成功比对上菌株的支架数据,便从长读长数据库中移除,以避免重复使用,并降低填补运算的计算量。
17、相比于现有技术,本发明的有益效果如下:
18、(1)现有技术中,二代测序由于读长短,菌株的基因组很难成环,测序完整度不高;三代测序碱基的错误率较高,价格较为昂贵。因此,本发明相较于二代测序方法,具有测序完整度高的优势,相较于三代测序方法,具有碱基测序质量高、成本低廉的优势。传统二代-三代混合测序的质量和完整度都较高,但费用非常昂贵。本发明采用单菌二代测序、混菌三代测序的策略,在保证基因组质量和完整度的前提下,大幅降低测序成本,单个菌株的测序费用为传统二代-三代混合测序方法的四分之一以下。
19、(2)二代测序获得的基因组草图中的缺失序列可以用一代测序和三代测序填补。本发明采用三代测序填补缺失序列的方法,并通过dna混合样品的方法降低单个菌株的测序成本,具有操作简便、成本低廉的优势。
20、(3)为了获得较高完整度的基因组序列,二代测序的单菌株数据量通常需要20gb及以上,三代测序的单菌株数据量通常为3gb及以上。本发明结合了二代测序和三代测序获得的序列,对所需的数据量进行了准确的估算,二代测序数据量仅需6.0gb,oxfordnanopore三代测序数据量仅需0.05m~0.1m gb(其中m为待分析菌株个数)。因此,相比于传统测序分析方法,本发明所需数据量低,具有测序成本低、序列分析快的优点。
技术特征:
1.一种结合二代和三代测序的多菌株全基因组分析方法,其特征在于,步骤包括:
2.根据权利要求1所述的结合二代和三代测序的多菌株全基因组分析方法,其特征在于,所述二代测序采用illumina二代测序,所述三代测序采用oxford nanopore三代测序。
3.根据权利要求1所述的结合二代和三代测序的多菌株全基因组分析方法,其特征在于,s1中所述dna浓度采用生物检测分析仪检测。
4.根据权利要求2所述的结合二代和三代测序的多菌株全基因组分析方法,其特征在于,每个菌株的illumina二代测序数据量为6.0gb。
5.根据权利要求2所述的结合二代和三代测序的多菌株全基因组分析方法,其特征在于,illumina二代测序得到的单独样品下机数据使用fastqc软件进行首次质量控制,使用fastp软件进行过滤,再次使用fastqc软件进行二次质量控制,得到illumina二代测序读长序列;使用metaspades软件,将illumina二代测序获得的读长序列组装成菌株支架数据。
6.根据权利要求2所述的结合二代和三代测序的多菌株全基因组分析方法,其特征在于,oxford nanopore三代测序数据量范围为(0.05~0.1)×m gb,其中m为dna混合样品中待分析的菌株数量。
7.根据权利要求2所述的结合二代和三代测序的多菌株全基因组分析方法,其特征在于,oxford nanopore三代测序得到的混合样本下机数据使用nanoplot软件进行首次质量控制,使用filtlong软件进行数据过滤,再次使用nanoplot软件进行二次质量控制。
8.根据权利要求2所述的结合二代和三代测序的多菌株全基因组分析方法,其特征在于,s2中所述的混合样品有效数据进行修剪和组装的过程采用flye软件。
9.根据权利要求2所述的结合二代和三代测序的多菌株全基因组分析方法,其特征在于,三代测序组装dna混合样品的长读长序列过程使用flye软件。
10.根据权利要求2所述的结合二代和三代测序的多菌株全基因组分析方法,其特征在于,所述填补过程使用unicycler软件,利用oxford nanopore三代测序产生的长读长序列,对illumina二代测序生成的每个菌株的支架数据逐一进行拼装和缺失序列填补,提高基因组的组装质量和连续性;在填补过程中,长读长序列一旦成功比对上菌株的支架数据,便从长读长数据库中移除,以避免重复使用,并降低填补运算的计算量。
技术总结
本发明公开了一种结合二代和三代测序的多菌株全基因组分析方法,属于环境微生物领域。本发明充分利用Illumina二代测序高通量、低成本的优势和Oxford Nanopore三代测序长读长、单分子测序的特点,提供了一种廉价、准确、高效的多菌株全基因组分析方法。首先,对每个菌株单独进行Illumina二代测序,利用MetaSPAdes软件对序列进行拼装,获得菌株的基因组草图。然后,对所有菌株的混合样品进行Oxford Nanopore三代测序。最后,利用Unicycler软件和混合样品的三代测序结果,对二代测序获得的各菌株基因组草图中的缺失序列进行填补,从而获得完成的、封闭成环的菌株基因组完成图。本发明具有成本低廉、操作简便、序列质量高等优点,适用于多菌株的全基因组测序分析。
技术研发人员:何崭飞,谷涵远,沈佳泉,杨莹莉,李群群,潘响亮
受保护的技术使用者:浙江工业大学
技术研发日:
技术公布日:2024/1/16
- 还没有人留言评论。精彩留言会获得点赞!