一种TH-302衍生物及其制备方法
本发明属于药物化学领域,具体涉及一种th-302衍生物及其制备方法。
背景技术:
1、艾伏磷酰胺(th-302)为n,n'-双(2-溴乙基)二氨基膦酸(1-甲基-2-硝基-1h-咪唑-5-基)甲酯,英文名是evofosfamide,th-302最初由threshold制药公司开发,是一种第二代hap,是由2-硝基咪唑部分连接到溴异磷酰胺芥末(br-ipm)组成的。硝基咪唑在细胞和体内系统中都要经历缺氧选择性生化还原,因此广泛应用于生物还原前药包括厌氧抗菌治疗以及抗癌症药物等药物的开发。溴异磷酰胺芥末(br-ipm)会与dna交联释放细胞毒素,发挥细胞毒作用,它在临床中被证明对抗肿瘤有着很好的疗效。将硝基咪唑的生物还原原理和溴异磷酰胺芥末的细胞毒素相结合便得到了著名的抗癌治疗药物艾伏磷酰胺(th-302)。
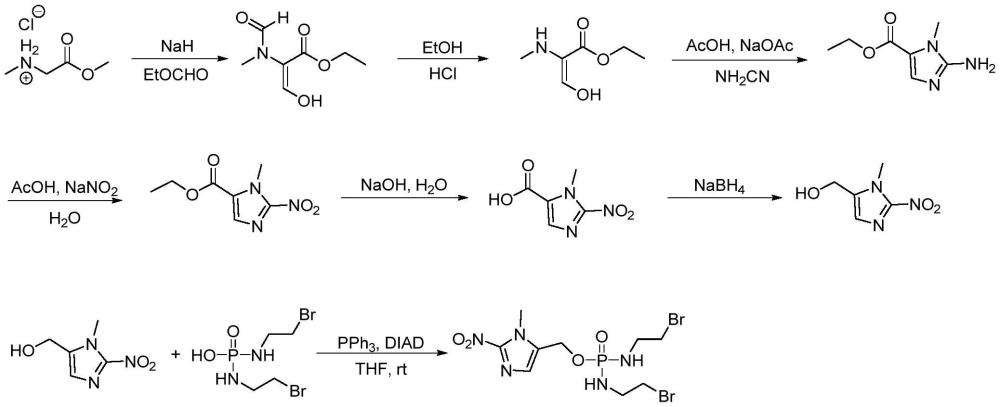
2、(1-甲基-2-硝基-1h-咪唑-5-基)甲醇传统的合成方法是以肌氨酸甲酯盐酸盐为初始原料,用氢化钠和甲酸乙酯处理,在α-碳和仲胺上发生反应得到(e)-3-羟基-2-(n-甲基甲酰胺)丙烯酸甲酯,然后在酸性乙醇中回流加热去除仲胺上的甲酰基,得到(e)-3-羟基-2-(甲基氨基)丙烯酸乙酯,随后在醋酸水溶液缓冲条件下回流用氰胺处理得到1-甲基-2-氨基-1h-咪唑-5-甲酸乙酯,最后在醋酸水溶液和亚硝酸钠作用下生成(1-甲基-2-硝基-1h-咪唑-5-基)甲醇,然后与n,n'-双(2-溴乙基)二氨基膦酸通过mitsunobu反应合成th-302,其中,文献m.matteucci,j.x.duan,h.jiao.phosphoramidate alkylator prodrugs[p].us invention patents,public,wo2007002931,2007.公开了其合成方法。
3、
4、该方法以肌氨酸甲酯盐酸盐为初始原料会导致合成的(1-甲基-2-硝基-1h-咪唑-5-基)甲醇的氮原子上只能为甲基取代,合成方法局限,不利于后续各种药物分子的结构修饰。
技术实现思路
1、为了改变传统路线的局限性,本发明的目的是提供一种th-302衍生物及其制备方法,通过对th-302咪唑环氮原子上取代基的修饰来改变th-302的生物还原活性。
2、本发明提供的th-302衍生物,通式如下化合物i、ii:
3、
4、其中,r为甲基、乙基、丙基、异丙基中的一种。
5、化合物i、ii选自式ia-id、iia-iid所示的化合物:
6、
7、th-302衍生物的制备方法,步骤如下:
8、(1)缩合环化:在反应瓶中依次加入3-溴丙酮酸乙酯,2-氨基嘧啶和乙醇,升温至75℃搅拌反应16h。冷却至室温,减压浓缩后,加入二氯甲烷和饱和碳酸氢钠水溶液使其溶解,分液,用饱和碳酸氢钠水溶液洗涤有机相两次,分液后再用二氯甲烷萃取水相三次,合并有机相,用无水硫酸钠干燥,减压浓缩后得到黑色油状液体,加入乙酸乙酯并搅拌后析出固体,过滤,用乙酸乙酯洗涤并干燥得到咪唑[1,2-a]嘧啶-2-甲酸乙酯;滤液减压浓缩经硅胶柱层析[洗脱剂:v(石油醚)/v(乙酸乙酯)]纯化得到咪唑[1,2-a]嘧啶-3-甲酸乙酯;
9、(2)肼解:在反应瓶中加入咪唑[1,2-a]嘧啶-3-甲酸乙酯,乙醇和水合肼;反应混合物升温至75℃,加热搅拌反应16h。停止反应后,减压浓缩得到黄色固体,用乙醚洗涤后过滤,滤饼干燥得到2-氨基-1h-咪唑-5-甲酸乙酯;
10、(3)氧化:在反应瓶中加入亚硝酸钠和水,搅拌使其溶解,在-8℃时缓慢滴加溶有2-氨基-1h-咪唑-5-甲酸乙酯的乙酸溶液,滴加完反应12h。停止反应后,用二氯甲烷萃取三次,合并有机相,用无水硫酸钠干燥,减压浓缩后经硅胶柱层析[洗脱剂:v(石油醚)/v(乙酸乙酯)]纯化得到2-硝基-1h-咪唑-5-甲酸乙酯;
11、(4)碘代:向反应瓶中加入2-硝基-1h-咪唑-5-甲酸乙酯,碘代试剂(碘甲烷、碘乙烷、1-碘丙烷、2-碘代丙烷),碳酸钾和乙腈,升温至65℃,加热搅拌反应15h;停止反应后,冷却至室温,过滤,滤液减压浓缩后用异丙醇洗涤并过滤,滤液减压浓缩后经硅胶柱层析[洗脱剂:v(石油醚)/v(乙酸乙酯)]分离纯化得到1-烷基-2-硝基-1h-咪唑-5-甲酸乙酯和1-烷基-2-硝基-1h-咪唑-4-甲酸乙酯;
12、实验过程中发现:随着取代基团的空间位阻效应增大,空间位阻作用相对更大的1-烷基-2-硝基-1h-咪唑-5-甲酸乙酯反而比1-烷基-2-硝基-1h-咪唑-4-甲酸乙酯更有利于生成,这说明该类结构稳定性的决定因素是电子效应而非位阻效应,即1-烷基-2-硝基-1h-咪唑-5-甲酸乙酯比1-烷基-2-硝基-1h-咪唑-4-甲酸乙酯具有更强的电子效应稳定性,随着取代基团的给电子能力增大,更加有利于1-烷基-2-硝基-1h-咪唑-5-甲酸乙酯的生成;
13、(5)还原:反应瓶中依次加入1-烷基-2-硝基-1h-咪唑-5-甲酸乙酯或1-烷基-2-硝基-1h-咪唑-4-甲酸乙酯,四氢呋喃,将反应体系冷却至0℃后向反应瓶中加入氢化铝锂,搅拌反应10min后,加热至0~65℃,搅拌反应6~18h;冷却至0℃,加入饱和硫酸铵溶液和氢氧化钠水溶液搅拌反应3h,过滤,滤液用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,减压浓缩经硅胶柱层析[洗脱剂:v(石油醚)/v(乙酸乙酯)]分离纯化得到(1-烷基-2-硝基-1h-咪唑-5-基)甲醇或(1-烷基-2-硝基-1h-咪唑-4-基)甲醇;
14、(6)取代:在氮气保护下向反应瓶中加入2-溴乙胺溴酸盐和无水二氯甲烷,将反应体系温度冷却至-78℃,向反应瓶中缓慢滴加三氯氧磷,滴毕,向反应瓶中滴加三乙胺和二氯甲烷的混合溶液,滴毕,保持-78℃继续搅拌反应1h,升温至25℃,继续搅拌反应2h;停止反应后,过滤,滤液浓缩后加入乙酸乙酯,再次过滤,滤液真空浓缩至黄色粘稠液体,加入四氢呋喃,-2℃滴加溴化钠水溶液,滴毕,保持-2℃搅拌15h,然后冷却至-20℃,冷冻析晶2h,过滤得白色固体,室温下自然烘干48h后得到n,n'-双(2-溴乙基)二氨基膦酸;
15、(7)mitsunobu反应:在氮气保护下向反应瓶中加入步骤(5)还原得到的(1-烷基-2-硝基-1h-咪唑-5-基)甲醇或(1-烷基-2-硝基-1h-咪唑-4-基)甲醇,步骤(6)得到的n,n'-双(2-溴乙基)二氨基膦酸,三苯基膦和无水四氢呋喃,冷却至0℃后,向反应瓶中滴加偶氮二羧酸二异丙酯(diad),升温至25℃,搅拌反应3h,停止反应后,减压浓缩后经硅胶柱层析[洗脱剂:v(乙酸乙酯)/v(甲醇)]纯化得到产物n,n'-双(2-溴乙基)二氨基膦酸(1-烷基-2-硝基-1h-咪唑-5-基)甲酯ia-id或n,n'-双(2-溴乙基)二氨基膦酸(1-烷基-2-硝基-1h-咪唑-4-基)甲酯。
16、本发明的有益效果体现在:本发明以2-氨基嘧啶和3-溴丙酮酸乙酯为原料,设计了一系列n,n'-双(2-溴乙基)二氨基膦酸(1-烷基-2-硝基-1h-咪唑-5-基)甲酯衍生物及其同分异构体n,n'-双(2-溴乙基)二氨基膦酸(1-烷基-2-硝基-1h-咪唑-4-基)甲酯衍生物的合成路线,该方法克服了传统合成路线中氮原子上取代基仅为甲基的局限性,一方面供电子取代基团的电子向正电荷部位转移,从而有效降低碳的正电性,从而使其稳定性增加,并且更利于后续双分子亲和取代反应的进行,另一方面也增加了合成化合物的选择性。
- 还没有人留言评论。精彩留言会获得点赞!