一种三并环类化合物及其制备方法和应用
本发明涉及化合物,更具体地,涉及一种三并环类化合物及其制备方法和应用。
背景技术:
1、免疫检查点阻断(icb)是通过免疫系统识别和清除肿瘤细胞的创新疗法,以程序性细胞死亡受体1/程序性细胞死亡配体1(pd-1/pd-l1)为代表的第一代免疫检查点抑制剂已逐渐成为肿瘤免疫(immuno-oncology,io)治疗的基石。在肿瘤免疫疗法领域进展最快的是pd-1/pd-l1抗体,其中o药(欧狄沃)和k药(可瑞达)在黑色素瘤和非小细胞肺癌的治疗方面展现出很好的疗效。然而,临床研究发现,pd-l1抗体的总体反应率较低(orr<30%)。同时大分子抗体药物的缺点也非常明显,首先,对相关组织和肿瘤细胞穿透性差,代谢半衰期长,只能通过注射给药等,其次,抗体药物具有免疫原性,因此会导致严重的不良反应,如免疫因子风暴综合征和致死性心肌炎等,而且,抗体药物制造和分离纯化过程很复杂,导致其生产成本非常高昂(o药,患者年治疗费用自费14万;k药,患者年治疗费用自费14万)。与大分子抗体药物相比,小分子化合物在药效动力学方面具有很多优势,例如,小分子化合物具有较好的口服生物利用度,对相关组织和肿瘤细胞渗透率高,制造成本低,半衰期合理等,因此研发小分子pd-l1调节剂,有望实现口服给药,降低患者用药经济负担,同时弥补抗体的缺陷,具有非常好的临床应用前景和现实意义。目前已报道的小分子化合物大多都是通过阻断pd-1/pd-l1相互作用,产生抗癌疗效,但是已报道的pd-l1小分子抑制剂口服生物利用度不高,需要进一步提高生物利用度(文献:j med chem.2021,64(11):7390-7403.doi:10.1021/acs.jmedchem.1c00010.),从而实现口服给药。
2、pd l1蛋白是一种重要的免疫抑制分子,有助于肿瘤免疫逃逸和肿瘤进展,其过表达可以传递负性调节信号,导致t细胞衰竭,因此通过设计合成pd-l1降解剂以降低或下调pd l1蛋白质含量,理论上可以进一步提高抗癌疗效。因为pd-l1降解剂能够促使pd-l1蛋白降解,这意味着pd-l1降解剂能够更彻底地解除pd-l1对t细胞的抑制作用,从而增强免疫系统对肿瘤的攻击能力,pd-l1降解剂可以更彻底的激活免疫系统。相比之下,pd-l1抑制剂主要通过阻止pd-l1与pd-1的结合来减轻抑制,而不一定能够消除pd-l1分子本身。进一步的,研究发现,pd-l1的过度表达与免疫逃逸有关,通过降低pd-l1蛋白质的含量,可以对抗这种上调的机制,有助于阻止肿瘤细胞逃避免疫监测。针对一些对传统治疗方法不敏感的肿瘤,pd-l1降解剂可提供一种新的治疗策略,通过增强免疫系统的攻击能力来改善治疗效果。另外,pd-l1降解剂具有更持久的治疗效果,因为它可以降解pd-l1分子,减少其在肿瘤细胞表面的表达,这有助于防止肿瘤细胞通过重新表达pd-l1来逃避免疫攻击,从而使治疗效果更加持久。目前国外科学家报道了溶酶体靶向嵌合体(lytacs)技术,可以有效降解pd-l1蛋白(文献:nature.2020,584(7820):291-297.doi:10.1038/s41586-020-2545-9.),但是lytacs分子量过大,不能口服给药。进一步科学研究发现,联苯类pd-l1小分子抑制剂可使pd-l1蛋白二聚化,随后发生内吞,通过溶酶体降解pd-l1蛋白(文献:j med chem.2022,65(5):3879-3893.doi:10.1021/acs.jmedchem.1c01682.),联苯类pd-l1小分子抑制剂结构的微小改变,可导致降解效率的巨大差异,这些信息提示,通过对联苯类pd-l1小分子抑制剂进行结构改造优化,有望获得可口服的,同时能有效降解pd l1的小分子降解剂,满足未满足的临床需求,造福广大肿瘤患者。
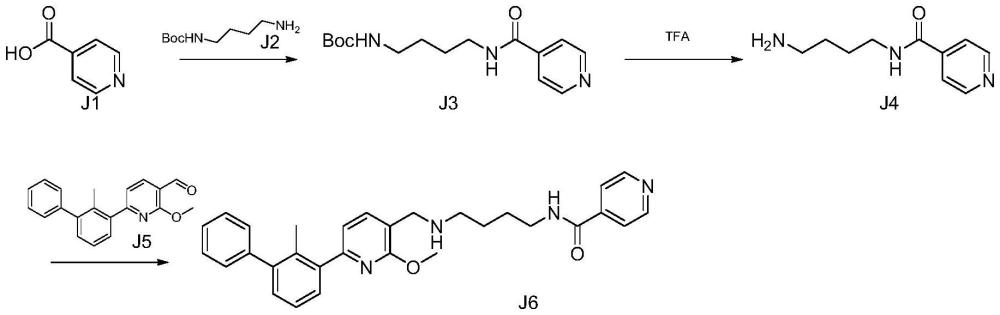
3、关于联苯类pd-l1小分子抑制剂的化学合成,专利(cn115417870a)报道了一条合成路线如下:
4、
5、申请人在重复该合成路线时,中间体j5的合成按照专利报道方法合成(如下所示,参考j med chem.2021 64(11),7390 7403中化合物23a的合成),收率不高,同时该路线使用到四氢铝锂、dmp等危险氧化还原试剂,后处理复杂,产生较多废液,不利于环保和工业化生产要求。
6、
7、参照专利合成方法,申请人发现第二步j3到j4采用tfa脱除保护基,容易生成副产物bp1同时中间体j4在酸性环境条件下,末端氨基容易成盐,氨基亲核性降低,导致第3步j5到j6还原胺化反应困难,目标产物j6收率不高,容易生成副产物bp2因此,除了需要发现全新的pd-l1降解剂外,仍需设计一条全新的三并环类pd-l1小分子抑制剂,以弥补现有技术的不足。
技术实现思路
1、本发明旨在至少解决上述现有技术中存在的技术问题之一,提出一种三并环类化合物及其制备方法和应用。
2、本发明提供的三并环类化合物,具有如下式(ⅰ)所示分子结构,能有效降低pd-l1蛋白,同时具有很高的口服生物利用度,有很好的临床应用前景。
3、本发明的第一方面提供一种三并环类化合物。
4、一种三并环类化合物,其特征在于,具有如下式(ⅰ)所示分子结构:
5、
6、其中,a选自
7、优选地,所述三并环类化合物具有如下式1-6所示任意一种结构式:
8、
9、
10、本发明的第二方面提供一种三并环类化合物的制备方法。
11、一种三并环类化合物的制备方法,包括如下步骤:
12、
13、其中,a选自
14、(1)先利用t1与t2发生suzuki偶联反应生成中间体t3;
15、(2)中间体t3和t4发生miyaura偶联反应生成中间体t5:
16、(3)中间体t5和t6发生suzuki偶联反应生成中间体t7;
17、(4)中间体t7与t8发生鲍奇还原反应生成中间体t9;
18、(5)中间体t9进一步和t10发生fmoc保护反应生成中间体t11;
19、(6)中间体t11在4m hcl的作用下脱掉boc保护基生成中间体t12;
20、(7)中间体t12和t13发生缩合反应生成中间体t14;
21、(8)中间体t14在20%哌啶的作用下脱掉fmoc保护基生成目标产物1-6。
22、优选地,步骤(1)中,所述suzuki偶联反应的温度为50-80℃,时间为1-10小时。
23、优选地,步骤(2)中,所述miyaura偶联反应反应的温度为70-90℃,时间为1-10小时。
24、优选地,步骤(3)中,所述suzuki偶联反应的温度为80-100℃,时间为1-10小时。
25、优选地,步骤(4)中,所述鲍奇还原反应采用还原试剂,所述还原试剂包括氰基硼氢化钠、醋酸硼氢化钠、硼氢化钠中的至少一种。
26、优选地,步骤(5)中,上fmoc保护基,反应条件需要加有机碱,所述有机碱包括三乙胺、dipea中的至少一种。
27、优选地,步骤(6)中,脱除boc保护基,反应需要在酸性条件下进行,所述酸性条件保护4m hcl/乙酸乙酯、tfa、4m hcl/二氧六环中的至少一种。
28、优选地,步骤(7)中,所述缩合反应采用缩合剂,所述缩合剂包括2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯、1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐、n,n”-羰基二咪唑中的至少一种。
29、优选地,步骤(7)中,所述缩合反应的温度为20-30℃,时间为0.5-3小时。
30、优选地,步骤(8)中,脱除fmoc保护基,反应溶剂为dmf,时间为0.5-3小时。
31、本发明的第三方面提供一种三并类化合物的应用。
32、一种三并环类化合物在制备治疗癌症药物中的应用。
33、一种三并环类化合物在制备治疗与pd-1/pd-l1相关的疾病药物中的应用。
34、优选地,所述药物还包括药学上可接受的盐、载体和辅料。
35、相对于现有技术,本发明的有益效果如下:
36、本发明提供的一种三并环类化合物,具有式(ⅰ)所示分子结构,能显著降解pd-l1蛋白,同时具有很高的口服生物利用度,是一种可口服给药的pd-l1降解剂,效果显著。同时上述三并环类化合物的合成路线避免了四氢铝锂、dmp等危险氧化还原试剂,同时避免了现有技术路线目标产物收率不高,容易产生副产物的步骤,原子利用率高,三废产生较少,利于环保和工业化生产要求。
- 还没有人留言评论。精彩留言会获得点赞!