一种以三聚磷腈为核的星形有机蓝光材料及其制备方法与流程
本发明涉及化工新材料技术领域,具体涉及一种有机发光材料及制备方法。
背景技术:
有机发光材料可以低成本制备大面积的柔性器件。有机发光二极管(OLED)具有启动电压低、亮度高、响应速度快、发光效率高等特点,可以应用于新一代大面积彩色柔性显示屏和固体照明等领域。
有机发光材料是有机光电转换器件及其产业发展的关键因素。目前已经开发出一些OLED发光材料,主要集中在三芳胺体系,蒽类衍生物,咔唑衍生物,稀土金属配合物等一些经典化合物体系。但是,现有材料还存在着许多应用方面的问题,比如使用寿命,发光亮度,发光效率等,开发综合性能优良的发光材料是一个重要的科研方向。
在全彩色大面积柔性显示领域,有机红光和绿光材料种类较多,有机蓝光材料种类比较少。目前,有机蓝光材料在使用寿命、发光效率和光谱纯度方面,与红光和绿光有机发光材料还有较大差距,开发新型有机蓝光材料是推动相关技术不断发展和应用的源动力。
技术实现要素:
本发明的目的在于提供一种综合性能优良的三聚磷腈为核的有机蓝光材料。
本发明另一个目的在于提供一种简单易行的制备三聚磷腈为核的有机蓝光材料的方法。
为实现第一个目的,本发明的技术方案如下:
本发明提供了一种以三聚磷腈为核、蒽环为发光基团的超支化星形分子结构,中间以苯环或寡聚苯来实现环三磷腈核与发光基团的化学键连接,其特征在于,该三聚磷腈为核的有机发光材料通式是:
其中n=1,2;蒽环通过9位碳原子与苯环桥连,连接位置可以是苯环的对位、间位或邻位;R为蒽环上任何可能的取代基,可以是亲电基团、供电子基团、芳香基团或者烷基等。
为实现第二个目的,本发明还提供了具有上述新型结构的有机发光材料的制备方法,特点是包括两步反应:
步骤(1):将碳酸钾(K2CO3)、溴代苯酚、四氢呋喃(THF)按照一定比例混合、搅拌、得到白色悬浊液,加热恒温一段时间,悬浊液变澄清,然后加入六氯环三磷腈,在一定温度和搅拌条件下反应一段时间,直到出现白色固体;停止反应,将反应液过滤、用饱和食盐水萃取滤液,对有机层经过干燥、过滤、浓缩、沉淀、洗涤,干燥后得中间产物;
步骤(2):将上述步骤(1)所制备的中间产物、K2CO3溶液、蒽硼酸、四三苯基膦钯、THF等混合均匀,在氮气保护下,一定温度和搅拌条件下反应一段时间,直到反应液由浅黄色变为棕色;停止反应,过滤、沉淀、洗涤、干燥、精制,得粉末状固体。
相对于现有技术,本发明具有如下优点:
1.本发明设计了一种新型有机蓝光材料——以三聚磷腈为核、蒽环为发光基团的超支化星形分子。
2.本发明将三聚磷腈热稳定性好的特点与蒽的高蓝光纯度和高发光效率特点结合起来,以实现新型有机蓝光材料综合性能提升,热稳定性和发蓝光性能方面优势集成。
3.本发明提供的三聚磷腈为核的有机蓝光材料的制备方法简单易行,容易实现工业化生产。
附图说明
图1为实施例1所得的三聚磷腈为核的星形有机蓝光材料的紫外可见光谱(CH2Cl2溶解);
图2为实施例1所得的三聚磷腈为核的星形有机蓝光材料的荧光发射谱图(激发波长为367nm,狭缝5nm,CH2Cl2溶解);
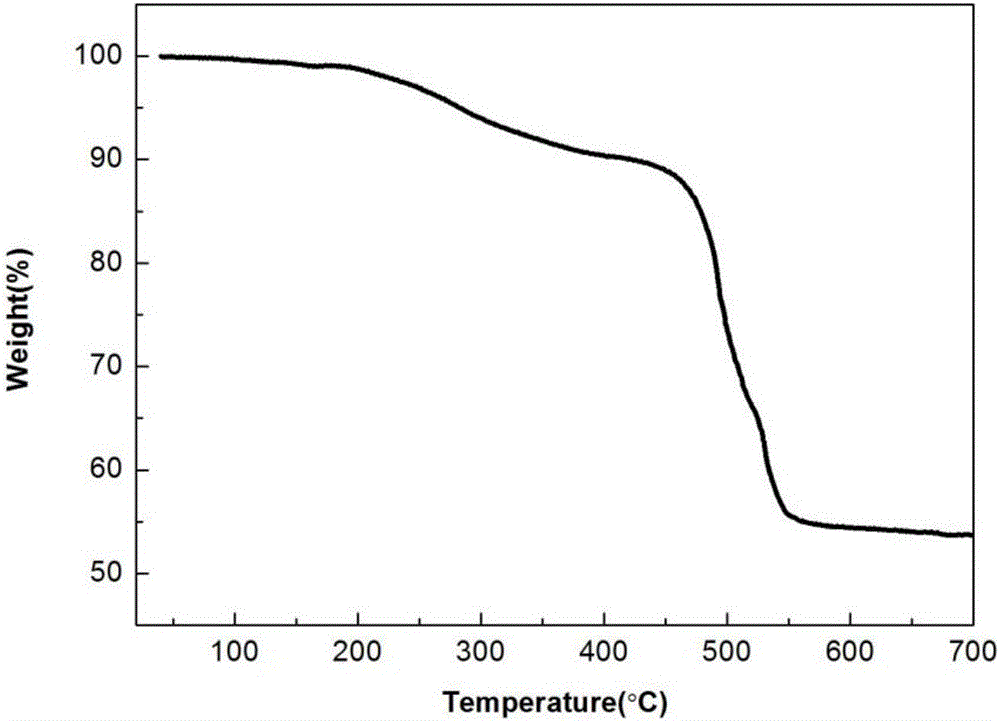
图3为实施例1所得的三聚磷腈为核的星形有机蓝光材料的热重分析谱图;
图4为实施例2所得的三聚磷腈为核的星形有机蓝光材料的紫外可见光谱(CH2Cl2溶解);
图5为实施例2所得的三聚磷腈为核的星形有机蓝光材料的荧光发射谱图(激发波长为367nm,狭缝5nm,CH2Cl2溶解);
图6为实施例2所得的三聚磷腈为核的星形有机蓝光材料的热重分析谱图。
具体实施方式
下面对本发明作进一步说明:
实施案例
实施例1:化合物001的合成,具体制备操作步骤如下:
步骤(1)将4.80g K2CO3(34.8mmol),5.00g对溴苯酚(29.0mmol)以及50mL干燥的THF溶液依次加入到两口烧瓶形成白色悬浊液,70℃恒温搅拌1小时,待悬浊液变澄清时,滴入1.00g六氯环三磷腈(2.90mmol)的THF稀溶液,70℃恒温反应48h,出现白色固体停止反应。将反应液过滤,滤掉固体,取滤液,然后用饱和NaCl溶液萃取3次,用无水MgSO4干燥有机层,过滤旋蒸,用甲醇洗涤2次、过滤干燥,得中间产物3.14g,产率为92.7%。
步骤(2)将1.170g步骤(1)制备的上述中间产物(1.00mmol),2M K2CO3溶液5mL,含2.00g蒽硼酸(9.00mmol)的无水乙醇溶液,0.310g四三苯基膦钯(0.27mol),50mL THF加入到两口瓶中,反应液呈浅黄色,抽真空-充氮气连续换气3次。在90℃油浴的条件下,连续搅拌48h。当反应液由浅黄色变为棕色时停止反应。待反应液冷却至室温后过滤,将滤液倒入过量甲醇中,出现浅黄色固体,过滤,干燥,得粉末状固体,柱层析分离,淋洗剂为二氯甲烷/石油醚,获得目标产物1.170g,产率为66.8%。化合物001结构式是
核磁与质谱数据如下所示,1H NMR(400MHz,CDCl3)δ 8.43(s,6H),7.97(s,12H),7.66(d,J=13.8Hz,24H),7.48(s,12H),7.29(d,J=21.7Hz,12H),7.20–6.80(m,12H).
MALDI-TOF-MS(m/z):1751.9
实施例2:化合物002的合成,具体制备操作步骤如下:
步骤(1)将4.80g K2CO3(34.8mmol),5.00g三溴苯酚(29.0mmol)以及50mL干燥的THF溶液依次加入到两口烧瓶形成白色悬浊液,70℃恒温搅拌1小时,待悬浊液变澄清时,将1.00g六氯环三磷腈(2.90mmol)的THF稀溶液加入澄清液中,70℃恒温反应48h,出现白色固体时停止反应。将反应液过滤,滤掉固体,取滤液,然后用饱和NaCl溶液萃取3次,用无水MgSO4干燥有机层,过滤旋蒸,用甲醇洗涤2次、过滤干燥,得3.30g中间产物2,产率为96.0%。
步骤(2)将1.170g步骤(1)制备的上述中间产物(1.00mmol),2M K2CO3溶液5mL,含2.00g蒽硼酸(9.00mmol)的无水乙醇溶液,0.310g四三苯基膦钯(0.27mol),50mL THF加入到两口瓶中,反应液呈浅黄色,抽真空-充氮气连续换气3次。在90℃油浴的条件下,连续搅拌48h。当反应液由浅黄色变为棕色时停止反应。待反应液冷却至室温后过滤,将滤液倒入过量甲醇中,出现浅黄色固体,过滤,干燥,得粉末状固体,柱层析分离,淋洗剂为二氯甲烷/石油醚,获得目标产物1.318g,产率为75.2%。化合物002结构式是
核磁数据如下所示,1H NMR(400MHz,CDCl3)δ 8.33(s,6H),7.86(d,J=7.6Hz,12H),7.44(d,J=8.0Hz,12H),7.23(d,J=27.2Hz,12H),6.96(dd,J=37.1,30.4Hz,36H).
MALDI-TOF-MS(m/z):1751.5
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求中。
- 还没有人留言评论。精彩留言会获得点赞!