基于蒽醌的二氯均三嗪型活性分散染料及其制备方法与流程
本发明涉及染料合成技术领域,尤其涉及一种基于蒽醌的二氯均三嗪型活性分散染料及其制备方法。
背景技术:
活性分散染料以分散染料为母体结构,并带有活性基,兼具分散染料和活性染料的性质。第一只活性分散染料由英国ici公司于1958年推出。当时为解决锦纶织物上的染色问题,专门设计了名为procinyl的染料,该染料具有不同的母体结构和活性基,满足了多种纤维的染色需求。但是,随着活性分散染料的研究开发,人们逐渐发现这一类染料在对诸如涤/棉混纺织物染色时,很难做到均匀上染,应用性能较差,因而活性分散染料的发展受到了限制。
1988年,schollmeyer等人首次提出了超临界co2流体中纺织品的染色。由于该技术利用超临界co2流体代替水作为介质对纺织品进行染色,不消耗水也无废水产生,绿色环保,从而受到了人们的广泛关注。此后,超临界co2流体无水染色技术迅速发展。目前,超临界co2染色技术在合成纤维上的应用较为成熟,如涤纶、锦纶、丙纶及腈纶等,其染色性能优异,但是在天然纤维上的染色问题较多。这是由于天然纤维为极性纤维,在染色时,非极性的超临界co2无法打开足够多的氢键,使得染料(超临界染色常用染料为分散染料)难以扩散进入纤维内部,而且适用于天然纤维染色的酸性、活性和直接染料等在超临界co2流体中的溶解性差,适用于天然纤维染色的染料极少。
活性分散染料的运用使得天然纤维在超临界co2流体中染色困难的问题迎刃而解,该染料以分散染料为母体结构,在超临界co2中的溶解度较好,且染料分子中的活性基可以与天然纤维上的反应性基团发生反应,形成共价键结合,湿处理牢度优良。然而,活性分散染料现有的品种较少,色谱不全,无法满足天然纤维超临界co2染色的需求,如何使活性分散染料的合成方法多元化,增加活性分散染料类型变得至关重要。
技术实现要素:
为解决上述技术问题,本发明提供了一种基于蒽醌的二氯均三嗪型活性分散染料及其制备方法。本发明的制备方法,操作简单,反应条件较为温和,成本较低,反应效率高,应用性强,且获得的活性分散染料不仅可用于常规水浴染色,也可用于超临界co2流体无水染色。
本发明的一种基于蒽醌的二氯均三嗪型活性分散染料的制备方法,包括以下步骤:
(1)将1-氯蒽醌和n-(2-氨基乙基)苯胺或其衍生物溶于有机溶剂,在碱剂和铜系催化剂作用下,在保护气氛中于90-120℃下反应4-12h,得到式(i)所示的染料前驱体;
(2)式(i)所示的染料前驱体与三聚氯氰反应,得到式(ii)所示的基于蒽醌的二氯均三嗪型活性分散染料;其中,式(i)、式(ii)如下:
其中,r选自氢、甲基或甲氧基。
进一步地,在步骤(1)中,有机溶剂为n,n-二甲基甲酰胺、二甲基亚砜和甲苯中的一种或几种。
进一步地,在步骤(1)中,碱剂为碳酸钾、氢氧化钾和三乙胺中的一种或几种。
进一步地,在步骤(1)中,铜系催化剂为溴化亚铜、氯化亚铜、二水合氯化铜和铜粉中的一种或几种。
进一步地,保护气氛为氮气和/或氩气。
进一步地,在步骤(1)中,1-氯蒽醌、n-(2-氨基乙基)苯胺或其衍生物、碱剂和铜系催化剂的摩尔比为20:20-60:10-50:1-4。1-氯蒽醌和n-(2-氨基乙基)苯胺或其衍生物在碱剂和催化剂的作用下,发生ullmann偶联反应。
进一步地,在步骤(1)中,有机溶剂与1-氯蒽醌的用量比为2.5-7.5ml:1mmol。
进一步地,在步骤(1)之后,还包括对式(i)所示的染料前驱体进行纯化的步骤。
纯化时,先萃取出产物,浓缩、干燥后得到固体粉末,对固体粉末进行柱层析,得到纯化后的产物。
本发明还提供了一种采用上述方法所制备的基于蒽醌的二氯均三嗪型活性分散染料,其分子式如下:
当r为氢时,以下说明书中用ia表示式(i)所示的染料前驱体,由此得到的式(ii)的基于蒽醌的二氯均三嗪型活性分散染料用iia表示。当r为甲基时,用ib表示式(i)所示的染料前驱体,由此得到的式(ii)的基于蒽醌的二氯均三嗪型活性分散染料用iib表示。当r为甲氧基时,用ic表示式(i)所示的染料前驱体,由此得到的式(ii)的基于蒽醌的二氯均三嗪型活性分散染料用iic表示。
借由上述方案,本发明至少具有以下优点:
本发明的制备方法操作较为简单,且在反应过程中催化剂的用量少,反应条件较为温和,成本较低,反应效率高、应用性强。本发明提供的基于蒽醌的二氯均三嗪型活性分散染料,不仅可用于常规水浴染色,也可用于超临界co2流体无水染色。
上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例为例,并配合附图,详细说明如后。
附图说明
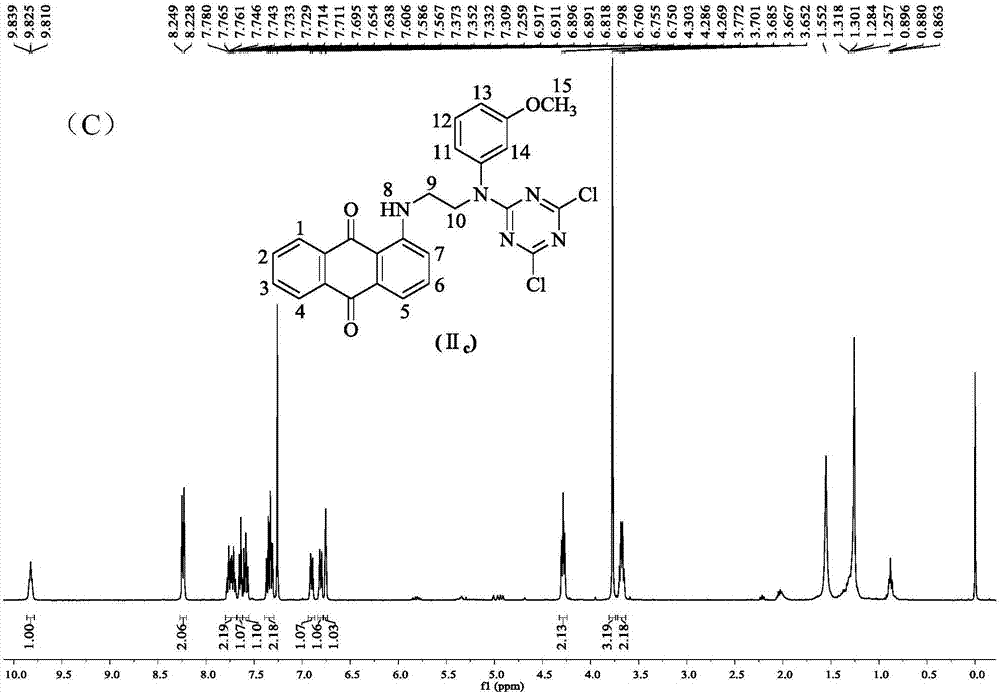
图1为基于蒽醌的二氯均三嗪型活性分散染料iia、iib和iic的核磁共振氢谱图;
图2为基于蒽醌的二氯均三嗪型活性分散染料iia、iib和iic在正己烷中的紫外-可见吸收光谱图;
图3为基于蒽醌的二氯均三嗪型活性分散染料iia对天然纤维(棉、真丝和羊毛)进行超临界co2染色的实物图。
具体实施方式
下面结合附图和实施例,对本发明的具体实施方式作进一步描述。以下实施例用于说明本发明,但不用来限制本发明的范围。
本发明中,n-(2-氨基乙基)-3-甲基苯胺和n-(2-氨基乙基)-3-甲氧基苯胺的合成方法如下:
在氮气氛围中,将间甲苯胺或3-甲氧基苯胺与2-溴乙胺氢溴酸盐在甲苯中回流反应20h,冷却后抽滤,并对滤渣进行柱层析提纯,使用丙酮作为洗脱剂,得到n-(2-氨基乙基)-3-甲基苯胺或n-(2-氨基乙基)3-甲氧基苯胺。
实施例1
本实施例提供了基于蒽醌的二氯均三嗪型活性分散染料iia及其制备方法,具体步骤如下:
(1)在100ml的三口烧瓶中依次加入0.002mol1-氯蒽醌、0.002moln-(2-氨基乙基)苯胺、0.002mol氢氧化钾和0.2mmol铜粉,在三口烧瓶中入通氮气,然后滴加5mln,n-二甲基甲酰胺,在100℃下反应10h。
(2)反应结束后冷却步骤(1)得到的反应液,然后以二氯甲烷为萃取剂萃取,浓缩,真空干燥,获得红棕色固体粉末。对该固体粉末进行柱层析提纯,洗脱剂为二氯甲烷与石油醚的混合物(体积比为1∶2),制得染料前驱体ia的提纯物,分离收率为50%;
(3)将0.001mol染料前驱体ia的提纯物和0.001mol碳酸钠分别溶解于15ml1,4-二氧六环和水中。将0.0015mol三聚氯氰溶解于5ml1,4-二氧六环中,并降温至0~5℃,向其中同时滴加上述配制的反应物溶液,在此温度下搅拌反应3h。反应完毕后,向反应液中加水,抽滤、水洗、真空干燥,获得染料iia的粗产物。然后对其进行柱层析提纯,洗脱剂为二氯甲烷与石油醚的混合物(体积比为2∶3),分离收率为60%。
利用核磁共振氢谱(400mhz,cdcl3)对本实施例制备的染料iia的提纯物进行结构表征,其结果如图1(a)所示。1hnmr(400mhz,cdcl3)δppm:9.84(t,j=5.4hz,1h,h-8),8.24(d,j=8.1hz,2h,h-1,h-4),7.74(dt,j=15.0,7.0hz,2h,h-2,h-3),7.65(d,j=7.0hz,1h,h-5),7.59(t,j=7.9hz,1h,h-6),7.47(t,j=7.6hz,2h,h-12,h-14),7.43-7.32(m,2h,h-7,h-13),7.24(d,j=7.6hz,2h,h-11,h-15),4.30(t,j=7.0hz,2h,h-10),3.66(dd,j=13.3,6.4hz,2h,h-9)。结果表明iia的各1h归属与理论结构相一致,说明通过本发明的方法得到了基于蒽醌的二氯均三嗪型活性分散染料iia。
称取微量染料iia的提纯物,溶于10ml正己烷中,利用tu-1810型紫外-可见吸收光谱仪测定其紫外-可见吸收光谱,结果见图2。经分析计算得知染料iia在正己烷中的最大吸收波长(λmax)为481nm,摩尔吸光系数为4.92×103l/(molcm)。
实施例2
本实施例提供了一种二氯均三嗪型蒽醌结构活性分散染料iib及其制备方法,具体步骤与实施例1类似。区别在于:
将步骤(1)中的n-(2-氨基乙基)苯胺换作n-(2-氨基乙基)-3-甲基苯胺;氢氧化钾、铜粉和n,n-二甲基甲酰胺的用量分别为0.004mol、0.1mmol和10ml,反应温度为90℃。其中n-(2-氨基乙基)-3-甲基苯胺的合成方法为:在氮气氛围中,将0.030mol间甲苯胺和0.010mol2-溴乙胺氢溴酸盐依次加入到100ml的三口烧瓶中,并滴加15ml甲苯,回流20h,冷却后抽滤,并对滤渣进行柱层析提纯,洗脱剂为丙酮,分离收率为80%。
在步骤(2)中进行柱层析提纯时,洗脱剂为二氯甲烷与石油醚的混合物(体积比为2∶3),制得染料前驱体ib的提纯物,分离收率为52%。
步骤(3)中的染料前驱体ia改为染料前驱体ib,经后处理后获得染料iib的粗产物,对其进行柱层析提纯,洗脱剂为二氯甲烷与石油醚的混合物(体积比为1∶1),分离收率为57%。
利用核磁共振氢谱(400mhz,cdcl3)对本实施例制备的染料iib的提纯物进行结构表征,其结果如图1(b)所示。1hnmr(400mhz,cdcl3)δppm:9.83(t,j=5.9hz,1h,h-8),8.24(dd,j=9.1,1.4hz,2h,h-1,h-4),7.74(dtd,j=20.0,7.4,1.3hz,2h,h-2,h-3),7.65(dd,j=7.3,1.0hz,1h,h-5),7.61-7.56(m,1h,h-6),7.38-7.30(m,2h,h-7,h-12),7.18(d,j=7.6hz,1h,h-13),7.02(d,j=6.1hz,2h,h-11,h-14),4.27(t,j=7.0hz,2h,h-10),3.72-3.63(m,2h,h-9),2.37(s,3h,h-15)。结果表明iib的各1h归属与理论结构相一致,说明通过本发明的方法得到了基于蒽醌的二氯均三嗪型活性分散染料iib。
称取微量染料iib的提纯物,溶于10ml正己烷中,利用tu-1810型紫外-可见吸收光谱仪测定其紫外-可见吸收光谱,结果见图2。经分析计算得知染料iib在正己烷中的最大吸收波长(λmax)为481nm,摩尔吸光系数为3.48×103l/(molcm)。
实施例3
本实施例提供了一种二氯均三嗪型蒽醌结构活性分散染料iic及其制备方法,具体步骤与实施例2类似。区别在于:
将步骤(1)中的n-(2-氨基乙基)-3-甲基苯胺换作n-(2-氨基乙基)-3-甲氧基苯胺,其合成方法为:在氮气氛围中,将0.025mol3-甲氧基苯胺和0.010mol2-溴乙胺氢溴酸盐依次加入到100ml的三口烧瓶中,并滴加20ml甲苯,回流20h,冷却后抽滤,并对滤渣进行柱层析提纯,洗脱剂为丙酮,分离收率为79%。
在步骤(2)中进行柱层析提纯时,洗脱剂为二氯甲烷与石油醚的混合物(体积比为1∶1),制得染料前驱体ic的提纯物,分离收率为48%。
步骤(3)中的染料前驱体ib改为染料前驱体ic,经后处理后获得染料iic的粗产物,对其进行柱层析提纯,洗脱剂为二氯甲烷与石油醚的混合物(体积比为3∶2),分离收率为55%。
利用核磁共振氢谱(400mhz,cdcl3)对本实施例制备的染料iic的提纯物进行结构表征,其结果如图1(c)所示。1hnmr(400mhz,cdcl3)δppm:9.82(t,j=5.7hz,1h,h-8),8.24(d,j=8.6hz,2h,h-1,h-4),7.74(dtd,j=13.6,7.6,6.3hz,2h,h-2,h-3),7.65(d,j=6.6hz,1h,h-5),7.59(t,j=7.9hz,1h,h-6),7.34(dd,j=16.8,8.5hz,2h,h-7,h-12),6.90(dd,j=8.3,2.1hz,1h,h-13),6.81(d,j=7.8hz,1h,h-11),6.76(t,j=2.1hz,1h,h-14),4.29(t,j=6.9hz,2h,h-10),3.77(s,3h,h-15),3.68(dd,j=13.3,6.4hz,2h,h-9)。结果表明iic的各1h归属与理论结构相一致,说明通过本发明的方法得到了基于蒽醌的二氯均三嗪型活性分散染料iic。
称取微量染料iic的提纯物,溶于10ml正己烷中,利用tu-1810型紫外-可见吸收光谱仪测定其紫外-可见吸收光谱,结果见图2。经分析计算得知染料iic在正己烷中的最大吸收波长(λmax)为482nm,摩尔吸光系数为2.48×103l/(molcm)。
实施例4
本实施例提供了染料前驱体ia的制备方法,该方法的步骤与实施例1的步骤(1)和(2)类似。区别在于:将步骤(1)中的氢氧化钾换作三乙胺,且用量为0.002mol;n,n-二甲基甲酰胺的用量为15ml。由此,在步骤(2)中染料前驱体ia的分离收率为30%。
实施例5
本实施例提供了染料前驱体ia的制备方法,该方法的步骤同实施例4。区别在于:将步骤(1)中的三乙胺换作氢氧化钾,且用量调整为0.002mol;将铜粉换作溴化亚铜,且用量调整为0.2mmol。由此,在步骤(2)中染料前驱体ia的分离收率为40%。
实施例6
本实施例提供了染料前驱体ia的制备方法,该方法的步骤同实施例5。区别在于:将步骤(1)中的溴化亚铜换作氯化亚铜,且用量为0.2mmol。由此,在步骤(2)中染料前驱体ia的分离收率为41%。
实施例7
本实施例提供了染料前驱体ia的制备方法,该方法的步骤同实施例5。区别在于:将步骤(1)中的溴化亚铜换作二水合氯化铜,且用量为0.2mmol。由此,在步骤(2)中染料前驱体ia的分离收率为37%。
实施例8
本实施例提供了染料前驱体ia的制备方法,该方法的步骤与实施例1的步骤(1)和(2)类似。区别在于:将步骤(1)中的氢氧化钾换作碳酸钾,且用量为0.002mol;有机溶剂n,n-二甲基甲酰胺改为二甲基亚砜,用量为15ml。由此,在步骤(2)中染料前驱体ia的分离收率为21%。
实施例9
本实施例提供了染料前驱体ia的制备方法,该方法的步骤同实施例8。区别在于:将步骤(1)中的有机溶剂二甲基亚砜换作甲苯,用量为15ml。由此,在步骤(2)中染料前驱体ia的分离收率为20%。
实施例10
本实施例提供了染料前驱体ia的制备方法,该方法的步骤与实施例1的步骤(1)和(2)类似。区别在于:将步骤(1)中的n-(2-氨基乙基)苯胺的用量调整为0.006mol,有机溶剂n,n-二甲基甲酰胺的用量调整为10ml。由此,步骤(2)中染料前驱体ia的分离收率为53%。
实施例11
本实施例提供了染料前驱体ia的制备方法,该方法的步骤与实施例1的步骤(1)和(2)类似。区别在于:步骤(1)中氢氧化钾和n,n-二甲基甲酰胺的用量分别为0.001mol和10ml。由此,步骤(2)中染料前驱体ia的分离收率为43%。
实施例12
本实施例提供了染料前驱体ia的制备方法,该方法的步骤与实施例1的步骤(1)和(2)类似。区别在于:步骤(1)中氢氧化钾的用量为0.005mol。步骤(2)中染料前驱体ia的分离收率为51%。
实施例13
本实施例提供了染料前驱体ia的制备方法,该方法的步骤与实施例1的步骤(1)和(2)类似。区别在于:步骤(1)中氢氧化钾、铜粉和n,n-二甲基甲酰胺的用量分别为0.004mol、0.4mmol和10ml。步骤(2)中染料前驱体ia的分离收率为54%。
实施例14
本实施例提供了染料前驱体ia的制备方法,该方法的步骤与实施例1的步骤(1)和(2)类似。区别在于:步骤(1)中铜粉的用量为0.1mmol,反应温度为120℃。步骤(2)中染料前驱体ia的分离收率为55%。
实施例15
本实施例提供了染料前驱体ia的制备方法,该方法的步骤与实施例14类似。区别在于:步骤(1)中的反应温度为90℃,反应时间为4h。步骤(2)中染料前驱体ia的分离收率为43%。
实施例16
本实施例提供了染料前驱体ia的制备方法,该方法的步骤与实施例15类似。区别在于:步骤(1)中的反应时间为12h。步骤(2)中染料前驱体ia的分离收率为59%。
由本发明的实施例1-3可制得基于蒽醌的二氯均三嗪型活性分散染料iia、iib和iic的提纯物。核磁共振氢谱(图1)的结果表明:三只染料的各1h归属与理论结构中的相一致。紫外-可见吸收光谱(图2)的分析结果显示:三只染料在非极性溶剂正己烷中的最大吸收波长(λmax)集中在480nm左右,而三者的摩尔吸光系数大小依次为iia>iib>iic。
由本发明实施例4-16的结果分析:各反应条件对染料前驱体ia的分离收率均有影响,尤其是碱剂种类、铜系催化剂种类和有机溶剂用量;适当控制各工艺参数,可使反应在较温和的条件下进行,如当铜系催化剂用量为原料的5%(以1-氯蒽醌的摩尔数计算),反应温度为90℃,时间为4h时,染料前驱体ia的分离收率也可在40%以上,反应效率高、应用性强。
此外,本发明制得的二氯均三嗪型蒽醌结构活性分散染料iia可成功应用于天然纤维(棉、真丝和羊毛)的超临界co2流体无水染色中,应用性能优异,如图3所示,其中(a)、(b)和(c)分别为棉、真丝和羊毛织物的超临界co2染色实物。对比图3结果可发现,三种染色织物的主色调均为红色,且在羊毛织物上的色泽最深。
以上所述仅是本发明的优选实施方式,并不用于限制本发明,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变型,这些改进和变型也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!