一种室温相变储能材料及其制备方法与流程
本发明涉及相变储能材料技术领域,特别是涉及一种室温相变储能材料及其制备方法。
背景技术:
我国青海盐湖地区镁盐储量极为丰富,且品质较高,但大量的镁盐因未找到合适的用途被公认为“镁害”。在众多镁盐中,mgcl2·6h2o具有高的熔化热(171kj/kg),已经作为相变储能材料被广泛研究,但由于相变温度较高(118℃),实现其在室温下应用,还必须与cacl2·6h2o等材料组成复合体系。然而,无机盐水合物应用在相变储能领域,普遍存在过冷度大、导热性差和相分离等问题。羟基化碳纳米管具有羟基基团,这可以与相变材料中的离子相结合,有助于改善碳纳米管与相变材料间的润湿性能;碳纳米管比表面积较大为相变材料提供更大的非均匀成核界面和更多的成核点,可以促进体系发生非均匀成核,降低成核势垒,进而降低过冷度;碳纳米管具有良好的导热性能,能在相变材料中形成性能优越的导热网络,从而提高体系的导热性能,使体系对温度更敏感,降低温度滞后性。根据“点阵匹配原理”,无机盐(锶盐)与mgcl2·6h2o-cacl2·6h2o体系的晶体结构和晶格参数接近,使表面能降低,对形核起到催化作用,进而降低过冷度。羟基化碳纳米管与无机盐同时加入mgcl2·6h2o-cacl2·6h2o相变材料中作为复合成核剂可以产生协同作用,共同促进相变材料成核,从而使体系过冷度显著降低。
技术实现要素:
本发明的目的是针对现有技术中存在的技术缺陷,而提供一种高导热、高储热室温相变储能材料的制备方法。
为实现本发明的目的所采用的技术方案是:
本发明通过如下技术方案予以实现。
本发明的一种室温相变储能材料的制备方法,包括以下步骤:
(1)分别称取cacl2·6h2o和mgcl2·6h2o晶体,加热,使混合物完全熔化;
(2)将步骤(2)配制的体系60~100℃下磁力搅拌1-3h,搅拌过程中加入增稠剂和羟基化碳纳米管,40~50℃超声分散1~2h,直至体系混合均匀;
(3)再称取srcl2·6h2o和srco3共同作为无机盐成核剂分别加入到步骤(3)配制的体系中,其中:60~100℃下磁力搅拌1-3h,40~50℃超声分散1~2h,最终得到mgcl2·6h2o-cacl2·6h2o室温相变储能材料;
其中:步骤(2)中羟基化碳纳米管作为成核剂加入质量分数为0~0.5%,即羟基化碳纳米管的质量在mgcl2·6h2o-cacl2·6h2o室温相变储能材料中占0~0.5%;
步骤(3)中srco3srcl2·6h2o和srco3加入总质量分数为,即srco3srcl2·6h2o和srco3的质量在mgcl2·6h2o-cacl2·6h2o室温相变储能材料中占0~5%。
优选的,所述步骤(1)中的cacl2·6h2o和mgcl2·6h2o晶体通过以下步骤制备:将无水氯化钙、无水氯化镁溶解于去离子水中,60~100℃配制成近饱和溶液,趁热进行抽滤提纯,室温下静置24h,抽滤去除液体,获得高纯度的cacl2·6h2o、mgcl2·6h2o晶体;
优选的,所述步骤(1)中的cacl2·6h2o和mgcl2·6h2o晶体质量比为(1:5)~(5:1)。
优选的,所述步骤(2)中的增稠剂为羟乙基纤维素或聚丙烯酰胺。
优选的,所述步骤(3)中的srcl2·6h2o和srco3质量比为(1:5)~(5:1)。
优选的,所述反应体系中cacl2·6h2o和mgcl2·6h2o晶体的加入量分别为71.81wt.%和23.94wt.%,即cacl2·6h2o和mgcl2·6h2o晶体的质量在mgcl2·6h2o-cacl2·6h2o室温相变储能材料中占71.81wt.%和23.94wt.%,羟基化碳纳米管、srco3和srcl2·6h2o加入量分别为0.25wt.%、1wt.%和3wt.%,即羟基化碳纳米管、srco3和srcl2·6h2o在mgcl2·6h2o-cacl2·6h2o室温相变储能材料中占0.25wt.%、1wt.%和3wt.%。
本发明的另一方面,还包括一种通过上述方法制备的mgcl2·6h2o-cacl2·6h2o室温相变材料。
优选的,所述mgcl2·6h2o-cacl2·6h2o室温相变材料中含有质量分数为0~0.5%的羟基化碳纳米管成核剂,和总质量分数为0~5%的srcl2·6h2o和srco3无机盐成核剂。
优选的,在所述mgcl2·6h2o-cacl2·6h2o室温相变材料中,cacl2·6h2o和mgcl2·6h2o晶体质量分数分别为71.81wt.%和23.94wt.%,,羟基化碳纳米管成核剂的质量分数为0.25wt.%,srco3的质量分数为1wt.%、srcl2·6h2o的质量分数为3wt.%。
优选的,所述mgcl2·6h2o-cacl2·6h2o室温相变材料相变温度范围为18.4~24.4℃,过
冷度变化范围为0.6~14.5℃,相变潜热变化范围为99.12~122.4j/g。
优选的,所述mgcl2·6h2o-cacl2·6h2o室温相变材料循环熔化-凝固50次后,相变潜热保持稳定。
与现有技术相比,本发明的有益效果是:
(1)由该方法制备的mgcl2·6h2o-cacl2·6h2o相变储能材料是应用性较好的室温相变储能材料体系,其合成工艺简单可行,可重复性强。
(2)由该方法将羟基化碳纳米管加入到mgcl2·6h2o-cacl2·6h2o相变材料,羟基化碳纳米管具有羟基基团,这可以与相变材料中的离子相结合,有助于改善碳纳米管与相变材料间的润湿性能,使碳纳米管可以较为长久的均匀分散在相变材料中,增加整个体系的稳定性。
(3)由该方法将羟基化碳纳米管加入到mgcl2·6h2o-cacl2·6h2o相变材料中,羟基化碳纳米管比表面积较大为相变材料提供更大的非均匀成核界面和更多的成核位点,可以促进体系发生非均匀成核,降低成核势垒,进而降低过冷度。采用温度记录仪对材料冷却曲线进行记录,当仅加入羟基化碳纳米管逐渐增加加入量,体系过冷度逐渐降低,当加入量为0.75g时,过冷度为9.1℃。
(4)碳纳米管具有良好的导热性能,能在相变材料中形成性能优越的导热网络,从而提高体系的导热性能,使体系对温度更敏感,降低了温度滞后性,提高了相变材料储热放热过程的能量利用效率。采用温度记录仪对材料冷却曲线进行记录,由图可知,在初始温度和降温条件基本相同的情况下,随着羟基化碳纳米管加入量逐渐增加,各组样品到达相变点的时间逐渐减小。
(5)羟基化碳纳米管与无机盐同时加入mgcl2·6h2o-cacl2·6h2o相变材料中作为复合成核剂可以产生协同效应,共同促进相变材料成核,从而使体系过冷度显著降低。采用温度记录仪对材料冷却曲线进行记录,当cnts、srco3、srcl2·6h2o的引入量分别为0.75g、0.3g和0.9g时,体系过冷度仅为0.6℃。
(6)采用羟基化碳纳米管-无机盐复合成核剂相比较不添加成核剂的材料,有更强的储热能力和很好的循环稳定性。如图2所示,引入0.75g碳纳米管-无机盐复合成核剂的相变材料的相变潜热为116.1j/g,进行熔化-凝固循环50次,如图3所示,相变潜热为113.5j/g。
附图说明
图1是实施例1所得到的相变储能体系的冷却曲线;
图2是实施例1所得到的相变储能体系的差示扫描量热曲线;
图3是实施例2所得到的相变储能体系的冷却曲线;
图4是实施例2所得到的相变储能体系的差示扫描量热曲线;
图5是实施例3所得到的相变储能体系的冷却曲线;
图6是实施例4所得到的相变储能材料的冷却曲线;
图7是实施例4所得到的相变储能材料的差示扫描量热曲线;
图8是实施例4所得到的相变储能材料进行熔化-凝固测试50次后得到的差示扫描量热曲线。
具体实施方式
以下结合附图和具体实施例对本发明作进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
实施例1(以羟基化碳纳米管作为成核剂)
(1)将无水氯化钙、无水氯化镁溶解于去离子水中,60~100℃配制成近饱和溶液,趁热进行抽滤提纯,室温下静置24h,抽滤去除液体,获得高纯度的cacl2·6h2o、mgcl2·6h2o晶体;
(2)分别称取22.5g和7.5g的cacl2·6h2o和mgcl2·6h2o晶体于烧杯中,加热至60~100℃,保持10min,使混合物完全熔化;
(3)将步骤(2)配制的体系60~100℃下磁力搅拌1-3h,搅拌过程中加入0.15g羟乙基纤维素作为增稠剂和0.75g羟基化碳纳米管作为成核剂,40~50℃超声分散1~2h,直至体系混合均匀,最终得到mgcl2·6h2o-cacl2·6h2o室温相变储能材料。
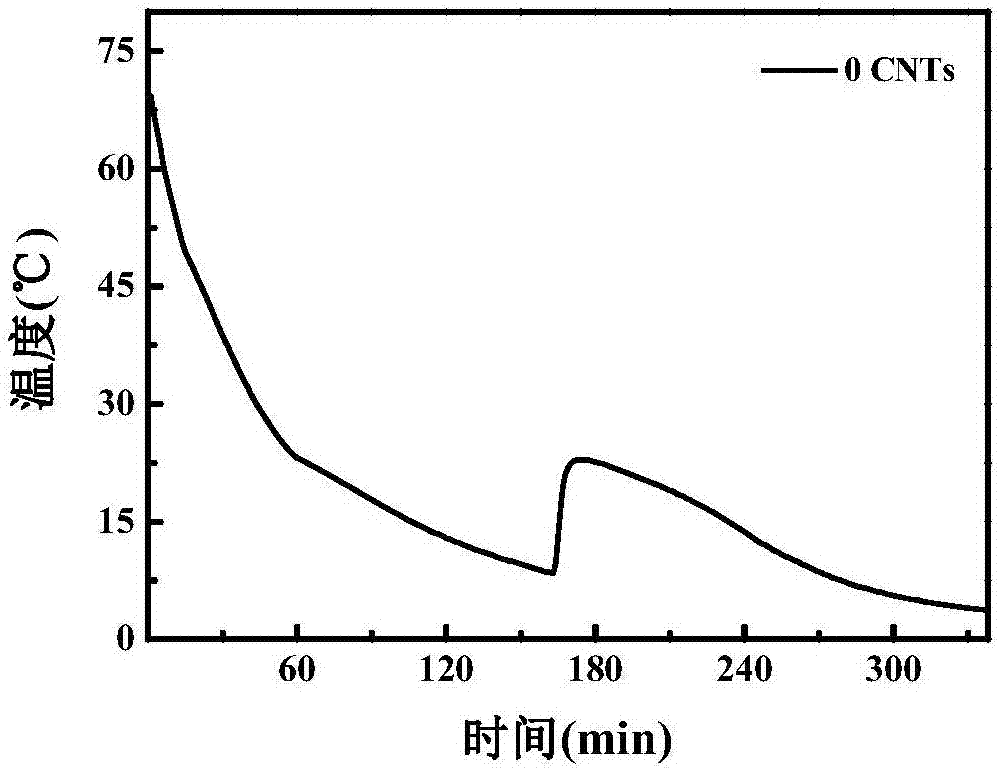
由图1可看出,实施例1得到的相变储能体系的过冷度为14.5℃,相变时间为165min,如图2可得,实施例1得到的相变储能体系的相变潜热为111.1j/g。
实施例2(以srcl2·6h2o、srco3作为成核剂)
(1)将无水氯化钙、无水氯化镁溶解于去离子水中,60~100℃配制成近饱和溶液,趁热进行抽滤提纯,室温下静置24h,抽滤去除液体,获得高纯度的cacl2·6h2o、mgcl2·6h2o晶体;
(2)分别称取22.5g和7.5g的cacl2·6h2o和mgcl2·6h2o晶体于烧杯中,加热至60~100℃,保持10min,使混合物完全熔化;
(3)将步骤(2)配制的体系高温下磁力搅拌2h,搅拌过程中加入0.15g增稠剂,高温超声分散1h,直至体系混合均匀;
(4)再称取0.9g的srcl2·6h2o和0.3g的srco3作为无机盐成核剂分别加入到步骤(3)配制的体系中,高温下磁力搅拌2h,高温超声分散1h,最终得到mgcl2·6h2o-cacl2·6h2o相变储能体系。
由图3可看出,实施例2得到的相变储能体系的过冷度为7.8℃,相变时间为71min,如图4可得,实施例2得到的相变储能体系的相变潜热为122.4j/g。
实施例3(以羟基化碳纳米管作为成核剂,并改变羟基化碳纳米管的加入量)
(1)将无水氯化钙、无水氯化镁溶解于去离子水中,60~100℃配制成近饱和溶液,趁热进行抽滤提纯,室温下静置24h,抽滤去除液体,获得高纯度的cacl2·6h2o、mgcl2·6h2o晶体;
(2)分别称取22.5g和7.5g的cacl2·6h2o和mgcl2·6h2o晶体于烧杯中,加热至60~100℃,保持10min,使混合物完全熔化;
(3)将步骤(2)配制的体系60~100℃下磁力搅拌1-3h,搅拌过程中加入0.15g羟乙基纤维素作为增稠剂和0.00g,0.15g,0.30g,0.45g,0.60g,0.75g羟基化碳纳米管作为成核剂,40~50℃超声分散1~2h,直至体系混合均匀,最终得到mgcl2·6h2o-cacl2·6h2o室温相变储能材料。
由图5可知,当加入0.00g,0.15g,0.30g,0.45g,0.60g,0.75g的cnts后,体系的过冷度逐渐减小,分别对应于15、14.5、13.6、9.4和9.1℃,可以看出随着羟基化碳管质量分数的增加,相变材料过冷度呈逐渐减小趋势,说明羟基化碳管起到了成核剂的作用。
实施例4
(1)将无水氯化钙、无水氯化镁溶解于去离子水中,60~100℃配制成近饱和溶液,趁热进行抽滤提纯,室温下静置24h,抽滤去除液体,获得高纯度的cacl2·6h2o、mgcl2·6h2o晶体;
(2)分别称取22.5g和7.5g的cacl2·6h2o和mgcl2·6h2o晶体于烧杯中,加热至60~100℃,保持10min,使混合物完全熔化;
(3)将步骤(2)配制的体系60~100℃下磁力搅拌1-3h,搅拌过程中加入0.15g羟乙基纤维素作为增稠剂和0.75g羟基化碳纳米管作为成核剂,40~50℃超声分散1~2h,直至体系混合均匀;
(4)再称取0.9g的srcl2·6h2o和0.3g的srco3作为无机盐成核剂分别加入到步骤(3)配制的体系中,60~100℃下磁力搅拌1-3h,40~50℃超声分散1~2h,最终得到mgcl2·6h2o-cacl2·6h2o室温相变储能材料,。
由图6可得实施例4得到的体系相变温度为19.9℃,过冷度为0.6℃,相变时间为41min。由图7可得实施例4得到的体系相变潜热为116.1j/g。
上述样品在没有外界干扰的情况下,进行熔化-凝固测试50次,由图8可知熔化-凝固50次后体系相变潜热为113.5j/g。
实施例5
(1)将无水氯化钙、无水氯化镁溶解于去离子水中,60~100℃配制成近饱和溶液,趁热进行抽滤提纯,室温下静置24h,抽滤去除液体,获得高纯度的cacl2·6h2o、mgcl2·6h2o晶体;
(2)分别称取22.5g和7.5g的cacl2·6h2o和mgcl2·6h2o晶体于烧杯中,加热至60~100℃,保持10min,使混合物完全熔化;
(3)将步骤(2)配制的体系60~100℃下磁力搅拌1-3h,搅拌过程中加入0.15g羧甲基纤维素作为增稠剂和0.75g羟基化碳纳米管作为成核剂,40~50℃超声分散1~2h,直至体系混合均匀;
(4)再称取0.9g的srcl2·6h2o和0.3g的srco3作为无机盐成核剂分别加入到步骤(3)配制的体系中,60~100℃下磁力搅拌1-3h,40~50℃超声分散1~2h,最终得到mgcl2·6h2o-cacl2·6h2o室温相变储能材料。
测试结果表明,实施例3所得到的mgcl2·6h2o-cacl2·6h2o室温相变储能材料在冷却过程中无法长时间保持均一稳定状态,有分层现象出现,所以在本体系中羧甲基纤维素并非一种适宜的增稠剂。
实施例6
(1)将无水氯化钙、无水氯化镁溶解于去离子水中,60~100℃配制成近饱和溶液,趁热进行抽滤提纯,室温下静置24h,抽滤去除液体,获得高纯度的cacl2·6h2o、mgcl2·6h2o晶体;
(2)分别称取22.5g和7.5g的cacl2·6h2o和mgcl2·6h2o晶体于烧杯中,加热至60~100℃,保持10min,使混合物完全熔化;
(3)将步骤(2)配制的体系60~100℃下磁力搅拌1-3h,搅拌过程中加入0.15g羟乙基纤维素作为增稠剂和0.45g羟基化碳纳米管作为成核剂,40~50℃超声分散1~2h,直至体系混合均匀;
(4)再称取0.9g的srcl2·6h2o和0.3g的srco3作为无机盐成核剂分别加入到步骤(3)配制的体系中,高温下磁力搅拌2h,40~50℃超声分散1~2h,最终得到mgcl2·6h2o-cacl2·6h2o室温相变储能材料。
实验结果表明,mgcl2·6h2o-cacl2·6h2o室温相变储能材料的相变温度为22.1℃,过冷度为5.8℃,相变时间为65min。
实施例7
(1)将无水氯化钙、无水氯化镁溶解于去离子水中,60~100℃配制成近饱和溶液,趁热进行抽滤提纯,室温下静置24h,抽滤去除液体,获得高纯度的cacl2·6h2o、mgcl2·6h2o晶体;
(2)分别称取22.5g和7.5g的cacl2·6h2o和mgcl2·6h2o晶体于烧杯中,加热至60~100℃,保持10min,使混合物完全熔化;
(3)将步骤(2)配制的体系60~100℃下磁力搅拌1-3h,搅拌过程中加入0.15g羟乙基纤维素作为增稠剂和0.5g羟基化碳纳米管作为成核剂,40~50℃超声分散1~2h,直至体系混合均匀;
(4)再称取0.75g的srcl2·6h2o和0.5g的srco3作为无机盐成核剂分别加入到步骤(3)配制的体系中,60~100℃下磁力搅拌1-3h,40~50℃超声分散1~2h,最终得到mgcl2·6h2o-cacl2·6h2o室温相变储能材料。
实验结果表明,mgcl2·6h2o-cacl2·6h2o室温相变储能材料的相变温度为20.3℃,过冷度为1.8℃,相变时间为54min。
实施例8
(1)将无水氯化钙、无水氯化镁溶解于去离子水中,60~100℃配制成近饱和溶液,趁热进行抽滤提纯,室温下静置24h,抽滤去除液体,获得高纯度的cacl2·6h2o、mgcl2·6h2o晶体;
(2)分别称取22.5g和7.5g的cacl2·6h2o和mgcl2·6h2o晶体于烧杯中,加热至60~100℃,保持10min,使混合物完全熔化;
(3)将步骤(2)配制的体系60~100℃下磁力搅拌1-3h,搅拌过程中加入0.15g羟乙基纤维素作为增稠剂和0.75g羟基化碳纳米管作为成核剂,40~50℃超声分散1~2h,直至体系混合均匀;
(4)再称取0.3g的srcl2·6h2o和0.1g的srco3作为无机盐成核剂分别加入到步骤(3)配制的体系中,60~100℃下磁力搅拌1-3h,40~50℃超声分散1~2h,最终得到mgcl2·6h2o-cacl2·6h2o室温相变储能材料。
实验结果表明,mgcl2·6h2o-cacl2·6h2o室温相变储能材料的相变温度为22.8℃,过冷度为7.1℃,相变时间为72min。
以上所述仅是本发明的优选实施方式,应当指出的是,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!