物质包封微囊及其制备方法与流程
本发明涉及新型物质包封微囊及其制备方法。
背景技术:
:众所周知,通过使一级结构被精密控制的高分子自组装,可以形成并获得微囊。由于这样的微囊能够多样化的分子设计,同时有呈现高分子固有性质之外新性能的可能性,因此研究了作为药物传递系统(DrugDeliverySystem:DDS)的载体、生物材料·功能材料等的使用。在专利文献1(日本特开平8-188541号公报)中,根据本发明人等的一部分人,公开了:具有不带电链段和带电链段的嵌段共聚物自组装而形成的静电结合型高分子胶束药物载体。在非专利文献1(SchlaadH.etal.,Macromolecules,2003,36(5),1417-1420)中,公开了:由聚(1,2-丁二烯)嵌段和聚(甲基丙烯酸铯)嵌段形成的嵌段共聚物与由聚苯乙烯嵌段和聚(1-甲基-4-乙烯基吡啶碘化物)嵌段形成的嵌段共聚物自组装而形成的被称为聚合物囊泡的微囊。在专利文献2(国际公开第2006/118260号小册子)中,根据本发明人等的一部分人,公开了:具有不带电亲水性链段和阳离子性链段的第1嵌段共聚物(例如PEG-聚阳离子等),与具有不带电荷的亲水性链段和阴离子段的第2嵌段共聚物(如PEG-聚阴离子等)自组装而形成的微囊。在非专利文献2(AnrakuY.etal.,J.Am.Chem.Soc.,2010,132(5),1631-1636)中,根据本发明者等的一部分人,公开了:具有不带电亲水性链段和带电链段的嵌段共聚物(例如PEG-聚阳离子等),和带与所述带电链段相反电荷的共聚物(如聚阴离子等)自组装而形成的微囊。通常认为由高分子的自组装而得到的所述各种微囊利用其空隙部包含、负载各种物质。(关于概论,参照非专利文献3(H.Nyinetal.SoftMatter,2006,2,940-949)以及非专利文献4(“脂质体应用的新开展”、秋吉一成等主编、NTS、2005年))。作为制备在空隙部中包封物质的微囊(以下有表示为“物质包封微囊”的情况)的方法,其代表为:将应该包封的物质(以下有表示为“被包封物质”的情况)与成为膜构成要素的高分子,或预先形成的高分子膜共同混合,经自组装,同时进行微囊的形成和向空隙部中的物质的封入的方法(以下有表示为“同时混合法”的场合)。作为具体例,可以列举出乳液聚合法(参照非专利文献5(F.Szoka,Jretal.,Proc.Natl.Acad.Sci.USA,197875(9)4194-4198))、脂质有机溶液滴落法(参照非专利文献6(Batzri,S.etal.,Biochim.BiophysActa1973,298,1015-1019))等。但是,在同时混合法中,有被包封物质的存在对由自组装而形成的微囊造成影响、阻碍微囊形成的情况,或即使微囊形成但在空隙部未包封物质的情况。此外,成膜时使用的有害有机溶剂的场合较多,也存在过程变得复杂,同时存在由有机溶剂造成的被包封物质易受损伤的问题。进而,难以形成具有均一的粒径、结构的微囊,为了确保这样的均一的粒径、结构不得不增加其他的工序,也有过程容易变得复杂的问题。因此,本方法通用性差,作为制造各种物质的包封微囊的方法没有实用性。另一方面,作为主要在向中空粒子中包封物质中使用的方法,存在将被包封物质导入现有中空粒子的空隙部之后,使其包封、负载的方法(以下有表示为[后负载法]的场合)(参照非专利文献7(W.Tongetal.J.Phys.Chem.B,2005,109,13159-13165)),也考虑将这样的方法应用于微囊。但是,在将后负载法应用于微囊的情况下,需要设法穿越空微囊的膜,将被包封物质导入空隙部。例如,可以想到将空微囊溶胀,使膜松弛,从产生的膜的间隙将被包封的物质渗透地导入空隙部后,使膜收缩并防止被包封物质脱落的方法,或在空微囊上形成孔,通过该孔将被包封物质导入空隙部后,封锁该孔并防止被包封物质脱落的方法等,然而任意一个都是极其复杂的,对于实用化非常地不利。此外,考虑在封入或负载被包封物质时,造成现有空微囊的粒径、结构紊乱的可能性高,非常没有实用性。另外,关于脂质体等的脂质双层膜微囊,也报告了在脂质双层膜上嵌入通道蛋白等的方法(参照非专利文献8(RanquinA,VerseesW,MiereW,SteyaertJ,GelderPV.TherapeuticNanoreactors:CombiningChemistryandBiologyinaNovelTriblockCopolymerDrugDeliverySystem.NanoLett.2005;5:2220-4)),然而过程仍然是极其复杂的,而且通用性极低,还是没有实用性。针对以上的
背景技术:
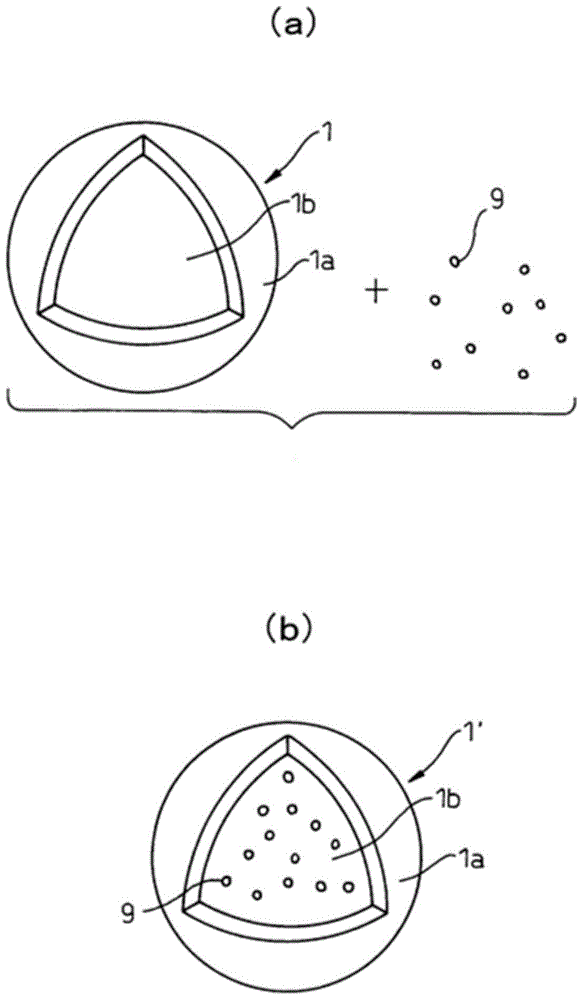
,本发明者等提出了极其意外的见解:通过在空微囊应包封的目标物质存在下,在水性介质中混合同时具有膜和由膜包围形成的空隙部的空微囊的方法(相当上述“后负载法”),且通过第1及第2聚合物的自组装,能够简便且效率地制备在空隙部(内水相)中包封了目标物质的物质包封微囊,其中,所述膜为包含作为具有不带电亲水性嵌段以及第1带电嵌段的嵌段共聚物的第1聚合物,和具有带与第1电荷性嵌段相反电荷的第2电荷性嵌段的第2聚合物,并进行了专利申请(专利文献3:国际公开第2011/145745号小册子)。现有技术文献专利文献专利文献1:日本特开平8-188541号公报专利文献2:国际公开第2006/118260号小册子专利文献3:国际公开第2011/145745号小册子非专利文献非专利文献:1SchlaadH.etal.,Macromolecules,2003,36(5),1417-1420非专利文献:2AnrakuY.etal.,J.Am.Chem.Soc.,2010,132(5),1631-1636非专利文献:3H.Nyinetal.SoftMatter,2006,2,940-949非专利文献:4“脂质体应用的新开展”、秋吉一成、辻井薰主编、NTS、2005年非专利文献:5F.Szoka,Jr.etal.,Proc.Natl.Acad.Sci.USA,197875(9)4194-4198非专利文献:6Batzri,S.etal.,Biochim.BiophysActa1973,298,1015-1019非专利文献:7W.Tongetal.,J.Phys.Chem.B,2005,109,13159-13165非专利文献:8RanquinAetal.,NanoLett.2005,5:2220-4技术实现要素:发明要解决的课题然而,在后负载法等的现有方法中,水性介质中存在的目标物质的浓度超过一定浓度时,对于空微囊的单分散体,由于在维持其单分散性的同时,不能封入目标物质,因此形成的物质包封微囊成为多分散体。此外,由于目标物质向空微囊的封入,作为依赖于水性介质中存在的目标物质的浓度的概率事件而产生,在后负载法等现有方法,难以控制封入量地向空微囊中封入两种以上的目标物质。此外,通过现有方法制备的静电相互作用型微囊,在可包封物质的尺寸方面具有限制。具体而言,被包封物质的直径的上限,最大限定为2~30nm程度,无法获得包封更大尺寸(例如,超过30nm)粒子的微囊。此外,由于通常的静电相互作用型微囊具有半透膜的性质,即使被包封物质的直径的下限限定为数千分子量的程度,也极其难以对具有更小分子量的小分子化合物进行包封。此外,现有的静电相互作用型微囊的物质包封率,虽然也取决于被包封物质的物理化学性质,但是受微囊和被包封物质混合时的溶液中的浓度对应的概率论的水平的限制,因此极其难以以更高效率地向微囊中包封物质。特别是,在相对于微囊的被包封物质的尺寸比较大的情况下(例如对直径100nm程度的微囊包封10nm(十分之一)以上的直径的粒子的场合),包封率极其低,通常具有不仅大量的被包封物质未被包封剩余下来,而且空的微囊大量地残留的问题。此外,在通过现有的方法制备的静电相互作用型微囊中,由于是通过微囊(或成为其构成要素的聚合物)和被包封物质在水性介质中混合,将被包封物质包封到微囊中,因此,极其难以将对于水溶性介质溶解性低的物质(低水溶性物质)有效地包封在微囊中。此处,本发明的第一目的在于提供一种物质包封微囊的制造方法,即,封入了与现有方法相比高浓度(例如,在后负载法中阻碍物质包封微囊的单分散集合体形成的浓度)的目标物质的物质包封微囊的单分散集合体,以及,在水性介质中存在的目标物资的浓度即使为高浓度(例如,在后负载法中阻碍物质包封微囊的单分散体形成的浓度),也可以形成物质包封微囊的单分散集合体。此外,本发明的第二目的在于提供一种物质包封微囊的制造方法,即,将2种以上的目标物质控制其封入量地封入的物质包封微囊,以及,能够将2种以上物质控制封入量地封入方法。此外,本发明的第三目的在于提供一种物质包封微囊的制备方法,即,在通过高分子的自组装的静电相互作用型微囊中,与以往相比,能够有效地包封较大直径的粒子或较小型的小分子化合物,或与现有相比,能够提高物质的包封率。此外,本发明的第四目的在于提供一种物质包封微囊的制造方法,即,在通过高分子自组装的静电相互作用型微囊中,能够有效地将在水性介质中不溶性或低水溶性的物质包封在微囊中。解决课题的手段本发明的主旨如下。[1]一种所述物质包封交联微囊的单分散体,是含有交联膜和所述交联膜包围成的内水相的并且在所述内水相中包封有目标物质的物质包封交联微囊的单分散体,其中所述交联膜包含作为具有不带电亲水性嵌段和第1带电嵌段的嵌段共聚物的第1聚合物,以及具有带与所述第1带电嵌段相反电荷的第2带电嵌段的第2聚合物,并且由所述第1和/或第2聚合物交联而成;所述内水相中含有的目标物质的浓度为,在所述第1和/或第2聚合物未交联以及所述目标物质未包封下,将与所述物质包封交联微囊的单分散体不同的空的非交联微囊的单分散体,混合在含有与所述物质包封交联微囊的内水相同一浓度的所述目标物质以及水性介质的混合液中的情况下,阻碍包封所述目标物质的物质包封非交联微囊的单分散体的形成的浓度。[2]根据上述[1]中记载的物质包封交联微囊的单分散体,具有0.2以下的多分散指数。[3]根据上述[1]或[2]中记载的物质包封交联微囊的单分散体,所述目标物质的重均分子量为10000~40000,所述内水相中含有的所述目标物质的浓度超过5mg/mL。[4]根据上述[3]中记载的物质包封交联微囊的单分散体,所述第1和/或所述第2聚合物,是通过选自由阳离子基团间形成的交联键、阴离子基团间形成的交联键、以及阳离子基团和阴离子基团间形成的交联键的组成的组的1种或2种以上的交联键交联而成,所述交联键的形成比例为所述交联膜中含有的阳离子基团和/或阴离子基团的总摩尔数的35%以上。[5]一种所述物质包封交联微囊,是含有交联膜和由所述交联膜包围成的内水相的并且在所述内水相中包封有第1目标物质和比所述第1目标物质分子量小的第2目标物质的物质包封交联微囊,所述第1目标物质,相比所述第2目标物质不存在时所述第1目标物质包含在所述内水相的场合是稳定的。其中所述交联膜包含作为具有不带电亲水性嵌段和第1带电嵌段的嵌段共聚物的第1聚合物,以及具有带与所述第1带电嵌段相反电荷的第2带电嵌段的第2聚合物,并且由所述第1和/或第2聚合物交联而成;[6]根据上述[5]中记载的物质包封交联微囊,所述第2目标物质是拥挤试剂。[7]一种物质包封微囊的制备方法,包含将空的交联微囊的单分散体、混合在含有所述目标物质、以及水性介质的混合液,形成物质包封交联微囊的单分散体的工艺;其中所述空的交联微囊的单分散体含有交联膜和所述交联膜包围城的内水相,并且在所述内水相中未包封有目标物质;所述交联膜包含作为具有不带电亲水性嵌段和第1带电嵌段的嵌段共聚物的第1聚合物,以及具有带与所述第1带电嵌段相反电荷的第2带电嵌段的第2聚合物,并且由所述第1和/或第2聚合物交联而成;其中所述物质包封交联微囊的单分散体包含含有所述第1和所述第2聚合物的且由所述第1和/或所述第2聚合物交联而成的交联膜,和由所述交联膜包围成的内水相,并在所述内水相中包封有所述目标物质。[8]根据上述[7]中记载的方法,所述空的交联微囊的单分散体、所述物质包封交联微囊的单分散体、所述空的非交联微囊的单分散体、以及所述物质包封非交联微囊的单分散体具有0.2以下的多分散指数。[9]根据上述[7]或[8]中记载的方法,所述目标物质的重均分子量为10000~40000,所述混合液中含有的所述目标物质的浓度为超过5mg/mL。[10]根据上述[9]中记载的方法,所述空的交联微囊以及所述物质包封微囊中,所述第1和/或所述第2聚合物,通过选自由阳离子基团间形成交联键、阴离子基团间形成交联键、以及阳离子基团和阴离子基团间形成交联键组成的组中的1种或2种以上的交联键交联而成,形成的所述交联键的比例为,所述交联膜中含有的阳离子基团和/或阴离子基团的总摩尔数的35%以上。[11]根据上述[7]~[10]任一项中记载的方法,还包括使所述物质包封交联微囊的单分散体和能够与所述第1和/或所述第2聚合物反应的交联剂进行反应的工艺。[12]一种物质包封微囊的制备方法,包含将含有第1物质包封交联微囊,混合在含有比所述第1目标物质分子量小的第2目标物质、以及水性介质的混合液中,并形成所述第2物质包封交联微囊的工艺;其中所述第1物质包封交联微囊含有交联膜、和所述交联膜包围成的内水相,并在所述内水相中包封有第1目标物质,所述交联膜包含作为具有不带电亲水性嵌段和第1带电嵌段的嵌段共聚物的第1聚合物,以及具有带与所述第1带电嵌段相反电荷的第2带电嵌段的第2聚合物,并且由所述第1和/或第2聚合物交联而成;其中所述第2物质包封交联微囊包含含有所述第1和第2聚合物的并由所述第1和/或所述第2聚合物交联而成的交联膜,以及所述交联膜包围成的内水相,在所述内水相中包封有所述第1和第2目标物质。[13]根据上述[12]中记载的方法,所述第1物质包封交联微囊是其单分散体,所述第2物质包封交联微囊是其单分散体。[14]根据上述[13]中记载的方法,所述混合液中含有的所述第2目标物质的浓度为,在所述第1和/或第2聚合物未交联的情况下,将与所述第1物质包封交联微囊的单分散体不同的第1物质包封非交联微囊的单分散体,混合在所述混合液中的情况下,阻碍包封有所述第1以及所述第2目标物质的物质包封非交联微囊的单分散体形成的浓度。[15]根据上述[12]~[14]任一项中记载的方法,还包括将空的交联微囊,混合在含有所述第1目标物质、以及水性介质的混合液中,根据需要,使其与能够和所述第1和/或所述第2聚合物反应的交联剂反应,形成所述第1物质包封交联微囊的工艺;其中所述空的交联微囊含有交联膜、和所述交联膜包围成的内水相,在所述内水相中未包封有所述第1和所述第2的目标物质的任一项;其中所述交联膜含有所述第1和所述第2的聚合物,由所述第1和/或第2的聚合物交联而成。[16]根据上述[15]中记载的方法,所述空的交联微囊是其单分散体,所述第1物质包封交联微囊是其单分散体。[17]根据上述[16]中记载的方法,所述混合液中含有的所述第1目标物质的浓度为,在所述第1和/或第2聚合物未交联的情况下,将与所述空的包封交联微囊的单分散体不同的空的包封非交联微囊的单分散体混合在所述混合液中的情况下,阻碍包封有所述第1目标物质的物质包封非交联微囊的单分散体形成的浓度。[18]根据上述[12]~[17]任一项记载的方法,所述第2目标物质为拥挤试剂。[19]一种吸附材料包封微囊,包括由包含作为具有不带电亲水性嵌段以及第1带电嵌段的嵌段聚合物的第1聚合物,以及,具有带所述第1带电嵌段相反电荷的第2带电嵌段的第2聚合物的膜形成的微囊,和该微囊中包封的吸附材料颗粒,所述第1以及第2聚合物的至少一方吸附于所述所述吸附材料颗粒。[20]根据上述[19]中记载的吸附材料包封微囊,所述第1和/或第2聚合物是交联的。[21]根据上述[19]或[20]中记载的吸附材料包封微囊,所述吸附材料颗粒是二氧化硅粒子。[22]根据上述[19]~[21]任一项中记载的吸附材料包封微囊,所述吸附材料颗粒的平均粒径为40nm~10μm。[23]根据上述[19]~[22]任一项中记载的吸附材料包封微囊,对所述吸附材料颗粒进行表面处理。[24]根据上述[19]~[23]任一项中记载的吸附材料包封微囊,所述吸附材料颗粒能够吸附小分子化合物。[25]一种吸附材料包封微囊的制造方法,在由膜形成的微囊上包封有吸附材料颗粒;其中所述膜含有作为具有不带电亲水性嵌段和第1带电嵌段的嵌段共聚物的第1聚合物,以及具有带与所述第1带电嵌段相反电荷的第2带电嵌段的第2聚合物;含有:(a)将所述第1和第2聚合物的一方与所述吸附材料颗粒混合,吸附于所述吸附材料颗粒的工序;(b)将所述工序(a)的混合物与所述第1或第2聚合物的另一方继续混合,在所述吸附材料颗粒的周围形成由含有第1和第2聚合物的膜形成的微囊,制作吸附材料包封微囊的工序。[26]根据上述[25]中记载的方法,还包括(c):交联所述工序(b)的微囊中的所述第1和/或所述第2聚合物的工序。[27]根据上述[25]或[26]中记载的方法,所述吸附材料颗粒是二氧化硅颗粒。[28]根据上述[25]~[27]任一项中记载的方法,所述吸附材料颗粒的平均粒径为40nm~10μm。[29]根据上述[25]~[28]任一项中记载的方法,还包括对所述吸附材料颗粒进行表面处理的工序。[30]根据上述[25]~[29]任一项中记载的方法,小分子化合物能吸附于所述吸附材料颗粒。[31]一种物质包封微囊的制备方法,在由膜形成的微囊上包封有目标物质,其中所述膜含有作为具有不带电亲水性嵌段和第1带电嵌段的嵌段共聚物的第1聚合物,以及具有带与所述第1带电嵌段相反电荷的第2带电嵌段的第2聚合物;含有:(a)能够将相比所述目标物质水溶性高的前体转换为所述目标物质的酶,包封在含有所述第1以及第2聚合物的膜形成的微囊中,准备酶包封微囊的工艺,以及,(b)相比于所述前驱体,对于目标物质低溶解性的条件下,所述前体向所述酶包封微囊内渗透,通过所述酶,所述前体向所述目标物质转换,所述目标物质析出,包封在所述酶包封微囊,制备物质包封微囊的工艺。[32]根据上述[31]中记载的方法,在所述工艺(b)中,通过将所述酶包封微囊和前体的水溶液混合,使所述前体向所述酶包封微囊内渗透。[33]根据上述[32]中记载的方法,在所述工艺(b)之前,还包括交联所述酶包封微囊的所述第1和/或所述第2聚合物的工艺。[34]一种物质包封微囊,其是通过上述[31]~[33]任一项中记载的方法而制备。[35]一种低水溶性物质包封微囊,从比所述目标物质水溶性高的前体转变得到的低水溶性物质与能将所述前体转变成所述低水溶性物质的酶一起被包封于由膜形成的微囊中,其中所述膜含有作为具有不带电亲水性嵌段和第1带电嵌段的嵌段共聚物的第1聚合物,以及具有带与所述第1带电嵌段相反电荷的第2带电嵌段的第2聚合物。[36]根据上述[34]或[35]中记载的低水溶性物质包封微囊,所述低水溶性物质在以相对于内水相的超过所述低水溶性物质的溶解度的浓度下包封。[37]根据上述[34]~[36]任一项中记载的低水溶性物质包封微囊,所述第1和/或所述第2聚合物是交联的。[38]一种含有上述[1]~[4]任一项中记载的微囊的单分散体,和/或,上述[5]、[6]、[19]~[22]以及[34]~[37]任一项中记载的微囊的药物传递系统。[39]一种用于将药物递送至对象的方法,包括,(a)在由膜形成的微囊中,包封能够将所述药物的前体转换为所述药物的酶的酶包封微囊的准备工序,其中所述膜含有作为具有不带电亲水性嵌段和第1带电嵌段的嵌段共聚物的第1聚合物,以及具有带与所述第1带电嵌段相反电荷的第2带电嵌段的第2聚合物,以及,(b)在对象特定的部位,使所述前驱体向所述酶包封微囊内渗透,通过所述酶,将所述前驱体转换为所述药物,形成所述药物的工序。[40]根据上述[39]中记载的方法,相比于所述药物所述前驱体的水溶性低,所述步骤(b)中,相比于所述前驱体在所述药物表现出相对低的溶解性的条件下,所述前体向所述酶包封微囊内渗透。[41]根据上述[39]或[40]中记载的方法,所述工序(b)中,还包括使所述药物析出并包封在所述酶包封微囊中,形成药物包封微囊的工序。发明的效果根据本发明,第一,提供了一种物质包封微囊的制备方法,与现有方法相比,可以形成高浓度(例如,在后负载法中阻碍物质包封微囊的单分散体形成的浓度)的目标物质封入的物质包封微囊的单分散体,以及,即使在水性介质中存在的目标物资的浓度为高浓度(例如,在后负载法中阻碍物质包封微囊的单分散体形成的浓度),也可以形成物质包封微囊的单分散体。根据本发明,第二,提供了一种物质包封微囊的制备方法,能将2种以上目标物质控制其封入量地封入的物质包封微囊,以及,能够将2种以上物质控制封入量地封入。根据本发明,第三,提供了一种方法,在提供了包封相对于微囊大小比较大的吸附材料颗粒,具有从所未有的新构造的物质包封微囊(吸附材料包封微囊),同时使极其效率地制备这种的吸附材料包封微囊称为可能。根据本发明,第四,可以效率地制备在现有方法中难以包封水溶性低的目标物质的微囊。附图说明[图1]图1(a)以及(b)为用于说明本发明的方法的图。[图2]图2为用于说明空微囊结构的图。[图3]图3(a)以及(b)为用于说明空微囊的膜结构的一种方式的图。[图4]图4(a)以及(b)为用于说明空微囊的膜结构的其他方式的图。[图5A]图5A为表示实施例I-1制得的FITC-Dex40k包封交联微囊的透射电子显微镜(TEM)的图像的图。[图5B]图5B为表示对比例I-1(同时混合法)制得的FITC-Dex40k包封非交联微囊的透射电子显微镜(TEM)的图像的图。[图5C]图5C为表示对比例I-2(后负载法)制得的FITC-Dex40k包封非交联微囊的透射电子显微镜(TEM)的图像的图。[图6]图6为表示为了确认封入在微囊内部的FITC-Dex40k释药行为而实施的GPC(凝胶渗透色谱法)的测试结果的图。[图7]图7(a)为表示空微囊的GPC测试结果的图,图7(b)为表示实施例I-2制得的精制后的细胞色素c包封交联微囊的GPC测试结果的图。[图8]图8(a)为表示实施例I-2制得的细胞色素c包封交联微囊的吸收光谱的图,图8(b)为表示实施例I-2制得的细胞色素c以及FITC-Dex4k包封交联微囊的吸收光谱的图。[图9]图9为表示圆二色谱的测试结果的图。[图10(A)]图10(A)为表示实施例I-3以及对比例I-3制得的微囊基于AlPcS2a吸收色的比色实验的结果的图。[图10(B)]图10(B)为表示实施例I-3以及对比例I-3制得的微囊的吸收光谱的图。[图11]图11为表示添加5mM、10mM或20mM的NaCl后微囊的透射电子显微镜(TEM)图像的图。[图12]图12为表示实施例I-4制得的CD封入交联微囊的透射电子显微镜(TEM)图像的图。[图13]图13为表示实施例I-4制得的CD封入交联微囊中从5-FC到5-FU的转变速度的图表。[图14]图14为表示实施例I-4制得的CD封入交联微囊中从5-FC到5-FU的转变酶活性的稳定性的图表。[图15]图15(a)~(d)为表示使用了实施例I-4制得的CD封入交联微囊的治疗所产生的肿瘤生长抑制效果的图表。[图16]图16(a)~(d)为表示使用了实施例I-4制得的CD封入交联微囊的治疗所产生的副作用(体重减少)得图表。[图17]图17(a)~(d)为表示使用了实施例I-4制得的CD封入交联微囊的治疗所产生的生存数的图表。[图18]图18为表示实施例I-4制得的CD封入交联微囊的血中滞留性的图表。[图19]图19为表示含有实施例II-1制得的MSN包封交联微囊的溶液的粒径分布的图表。[图20]图20为实施例II-1制得的MSN包封交联微囊的TEM照片。[图21]图21为实施例II-1制得的MSN包封交联微囊的毛细管电泳的色谱图。[图22]图22为包含表示实施例II-2制得的MSN包封交联微囊溶液的粒径分布的图表。[图23]图23为实施例II-2制得的MSN包封交联微囊的TEM照片。[图24]图24为实施例II-2制得的MSN包封交联微囊的毛细管电泳的色谱图。[图25]图25为表示含有参考例II-1制得的MSN包封交联微囊的溶液的粒径分布的图表。[图26]图26为参考例II-1制得的MSN包封交联微囊的TEM照片。[图27]图27为参考例II-1制得的MSN包封交联微囊的毛细管电泳的色谱图。[图28]图28(a)为实施例II-3A中使用的未处理MSN包封微囊的TEM照片,图28(b)为实施例II-3A制得的胺化MSN包封微囊的TEM照片。[图29]图29(a)为实施例II-3B中使用的未处理MSN包封微囊的TEM照片,图29(b)为实施例II-3B制得的磺酰化MSN包封微囊的TEM照片。[图30]图30为实施例II-3B中使用的未处理MSN包封微囊以及制得的磺酰化MSN包封微囊的X射线分析光谱。[图31]图31为表示实施例II-4A制得的孟加拉玫瑰红吸附胺化MSN包封微囊中玫瑰红的释放特性的图表。[图32]图32为表示实施例II-4B制得的吉西他滨吸附磺酰化MSN包封微囊中吉西他滨的释放特性的图表。[图33]图33(a)以及(b)为表示实施例II-4B制得的吉西他滨吸附磺酰化MSN包封微囊的对于A549细胞的细胞摄取特性的图表。图33(a)表示在不存在台盼蓝时全细胞内Cy3荧光量的测试结果,图33(b)表示存在台盼蓝染色时活细胞内Cy3荧光量的测试结果。[图34]图33(a)以及(b)为表示实施例II-4B制得的吉西他滨吸附磺酰化MSN包封微囊的对于A549细胞的细胞杀伤效果的图表。图34(a)表示培养48小时后的结果,图34(b)表示培养72小时后的结果。[图35]图35(a)以及(b)为通过实施例II-4B制得的吉西他滨吸附磺酰化MSN包封微囊的对于C26细胞的细胞摄取特性图。图35(a)表示在不存在台盼蓝时全细胞内Cy3荧光量的测试结果,图35(b)表示存在台盼蓝染色时活细胞内Cy3荧光量的测试结果。[图36]图36(a)以及(b)为表示实施例II-4B制得的吉西他滨吸附磺酰化MSN包封微囊的对于C26细胞的细胞杀伤效果的图表。图36(a)表示培养48小时后的结果,图36(b)表示培养72小时后的结果。[图37]图37(a)以及(b为表示)为表示使用实施例II-4B制得的吉西他滨吸附磺酰化MSN包封微囊的肿瘤治疗效果的图表。(a)表示肿瘤增大抑制效果,(b)表示副作用(体重减少)降低效果。[图38]图38为表示实施例II-4B制得的吉西他滨吸附磺酰化MSN包封微囊的血中滞留性的图表。[图39]图39为表示实施例II-4B制得的吉西他滨吸附磺酰化MSN包封微囊的肿瘤累积性的图表。[图40]图40为表示含有实施例III-1制得的β-半乳糖苷酶包封交联微囊溶液的粒径分布的图表。[图41]图41为表示含有实施例III-1制得的靛蓝染料包封微囊溶液的粒径分布的图表。[图42]图42为实施例III-1制得的靛蓝染料包封微囊TEM照片。[图43]图43为实施例III-1制得的靛蓝染料包封微囊的高分辨率的TEM照片。[图44]图44为表示含有比较例III-1制得的交联空微囊溶液的粒径分布的图表。[图45]图45为表示含有比较例III-1制得的靛蓝染料产生后的空微囊溶液的粒径分布的图表。[图46]图46为表示比较例III-1制得的靛蓝染料产生后的空微囊的TEM照片。具体实施方式以下根据具体的实施方式对本发明进行详细地说明。但本发明绝不受限于以下的实施方式,能在适宜变更的基础上进行实施。[A]定义本说明书中所谓的微囊,是指具有单层结构的膜,和由所述膜包围成的空隙部(内水相)的基本结构体。本发明中,基团的名称或作为其一部分的“烷基”表示一价的脂肪族饱和烃基。除非另有说明,烷基可以是链状、环状,也可以是链状和环状的结合。链状烷基可以是直链状也可以是支链状。环状烷基可以是单环式也可以是复环式,复环式的情况可以是连接环、缩合环、螺环。本说明书中的基团的名称或作为其一部分的“烷氧基”表示所述的烷基与二价的氧原子的一方的结合成键而形成的基团。本说明书中的基团的名称或作为其一部分的“芳基”表示一价的芳香族烃基。除非另有说明,芳基可以使单环式也可以是复环式,在复环式的情况下可以是连接环、缩合环,也可以是螺环。本说明书中的基团碳原子数表示为例如“C1-12烷基”的方式。在这里,“C1-12”意味着该烷基的碳原子数为1~12个。本说明书中的“卤素原子”意味着氟原子、氯原子、溴原子、或碘原子。本说明书中的有的基团“可以被取代”意味着该基团具有的1个或2个以上的氢原子可以被1个或2个以上的取代基(2个以上时可以相同也可以不同)取代。取代基的最大数与该基团具有的结构以及能够取代的氢原子数有关,若是本领域技术人员能够容易地确定。本说明书中的“取代基”,除非另有说明,可以选自由卤素原子、芳基、羟基、氨基、羧基、氰基、甲酰基、二甲基缩醛甲酰基、二乙基缩醛甲酰基、C1-6烷氧羰基、C2-7酰胺基、硅氧基、三-(C1-6烷基)硅氧基(其中C1-6烷基可以相同也可以不同)以及甲硅烷氨基组成的组。本说明书中的有关各种微囊使用的用语“单分散体”意味着粒径分布狭窄的微囊集合体。粒径分布的宽窄,优选基于多分散指数(polydispersityindex:PDI)判断,单分散体具有优选0.2以下,更优选0.15以下,进一步优选0.1以下的分散指数。本说明书中的有关各种微囊使用的用语“多分散指数”意味着表示粒径分布广度的无量纲指标,用语“平均粒径”意味着依据散射光强度基准的调和平均粒径(直径)。多分散指数以及平均粒径都是通过动态光散射法(dynamiclightscattering)测定。动态光散射法能够依据JISZ8826:2005(粒径分析-光子相关光谱法(Particlesizeanalysis-Photoncorrelationspectroscopy))实施。JISZ8826:2005由于是对应于稀颗粒浓度分散液体的标准,为了实现即使在高浓度样品中也能够测定其多分散指数以及平均粒径,在JISZ8826:2005上可以增加适当的变更。例如,能够采用后向散射检测替代侧向散射检测。通过采用后向散射检测,能够排除或降低多重散射的影响,据此,即使是在高浓度的样品中,也能够测定其多分散指数以及平均粒径。作为通过动态光散射法用于测定多分散指数以及平均粒径的市售设备可以列出例如Malvern社制ZetasizerNano-ZS。该设备采用后向散射检测,在低浓度~高浓度的广泛浓度范围的样品中,能够测定其多分散指数以及平均粒径。[B]物质包封微囊及其制备方法首先,对作为本发明基础的物质包封微囊以及制备方法进行说明。有关本发明的各微囊及其制备方法的特征,将在章[C]~[E]中重新说明,但关于这些章中没有特别注明的事项,适用本章的记载。[B1:物质包封微囊的制备方法]如上所述,作为物质包封微囊的主要的制备方法可以列出,(i)将被包封物质、成为膜构成要素的高分子、或预先形成的高分子膜一同混合,同时进行通过自组装的微囊的形成和将物质封入空隙部的方法(同时混合法)和,(ii)将预先形成的空微囊和被包封物质混合,向空微囊的空隙部中导入被包封物质,从而进行包封、负载的方法(后负载法)。本发明中,有关各个微囊的制备条件,除非另有说明,能够使用含有同时混合法或后负载法的任意方法。但是,作为本发明人等开发的方法的后负载法(详细参照上述专利文献3(国际公开第2011/145745号公报))具有,能以简便的方法有效地将包封物质导入微囊内,导入前后也几乎不损害微囊的结构,而且能够包封在同时混合法中包封困难的带电荷物质等的种种优点,因此是令人满意的。下面,有关物质包封微囊的制备方法,将后负载法作为前提进行说明,但本发明中,物质包封微囊的制备方法并不限于后负载法,在不违反后述的各物质包封微囊的特征的限制下,能够使用包含同时混合法的其他任意的方法。此外,在使用后负载法以外的方法向微囊中包封物质的情况下,作为其具体的条件,能够比照适用后述的后负载法的条件。例如,在同时混合法的情况下,除了并不按照后负载法预先制备空微囊,将作为空微囊的材料的聚合物等与被包封物质一同混合以外,基本上可以使用和后负载法相同的条件。(B1-1:后负载法的概要)关于后负载法的概要,下面使用图1进行说明。然而,图1是模型图,本发明并不限定于图1。如图1(a)所示,准备具有由膜1a包围了的空隙部1b的特定构造的空微囊,将其和被包封物质9一同在水性介质中混合。据此,如图1(b)所示,被包封物质9穿过空微囊1的膜1a被导入到空隙部1b中,制得被包封物质9被包封在空隙部1b中的物质包封微囊1’。以下,对后负载法进行详细叙述,在章节中对空微囊及被包封物质的详细情况进行重新叙述,此处对其他的条件及顺序进行说明。(B1-2:空微囊以及被包封物质的混合)后负载法包含空微囊以及将被包封物质在水性介质中混合的工序(应当指出,此处具有将成为混合对象的在水性介质中含有空微囊以及被包封物质的液体,此后称为“混合对象液”的情况)。进行混合的方法没有特别的限制,在水性介质中施加外力的方法进行。即,排除了将空微囊以及被包封物质添加到水性介质中静置,使其自然扩散混合的方法(此后有称为“静置、扩散混合”的场合)。作为在水性介质中施加外力的混合方法的例子,可以列出搅拌、振荡、冲击等。作为搅拌的方法的例子,可以列出,将含有混合对象液的容器通过涡流混合器等回旋搅拌的方法,或将溶液通过搅拌翼等直接搅拌的方法等。作为振荡的方法的例子,可以列出,将含有对象混合液的容器通过振荡器等振荡的方法。作为冲击的方法的例子,可以列出,对混合对象液通过超声波照射等含有振动的各种给与冲击的方法。通过这样的混合,物质被包封在微囊空隙部中,可以制备物质包封微囊。通过混合形成物质包封微囊的原因不明确,但通过向水性介质施加外力,剪切应力作用到空微囊上(因此,可以将在水性介质中施加外力的混合称为剪切应力下的混合)。通过这样的剪切应力,空微囊的结构被扰乱,分解成大致均匀的小聚集体,可以认为在这些小聚集体再次自组装微囊均匀地再生的同时,水性介质中存在的被包封物质在微囊再生时,被包封在微囊内(这样的机制也可以在后述的参考例中推测,确认通过混合分解成的微囊小聚集体)。这是在通常状态下难以发生的现象,利用了这样现象的后负载法可以说是极其新颖的。混合的条件没有限制,但若考虑到上述的机制,优选在在水性介质中空微囊的结构被充分地扰乱的同时,扰乱后微囊的结构能够再生的程度的条件。一般以力作用于混合对象液全体的程度进行混合即可,优选以混合对象液全体成为大致均匀的程度进行混合。具体的混合条件因混合方法而不同,例如,在搅拌的情况下,最好以一般500rpm以上,优选1000rpm以上,另外,一般10000rpm以下,优选5000rpm以下的转速来进行。若转速过低,会有难以形成均质的物质包封微囊的情况。若转速过高,会有损伤、破坏微囊或被包封物质的情况。此外,通过涡流混合器的搅拌时间,因转速而不同,一般为60秒以上,优选为120秒以上,另外,一般为10分钟以内,优选为5分钟以内。当搅拌时间过短时,会有难以形成均质的物质包封微囊的情况。当搅拌时间过长时,会有损伤、破坏微囊或被包封物质的情况。作为使用其他的混合方法(通过搅拌翼的搅拌、通过振荡器的振荡、通过超声波照射的冲击等)的情况的具体的条件,与将根据涡流混合器的搅拌在上述的转速以及搅拌时间下进行并获得的同等程度的力作用于混合对象液的方式,适宜地调整条件即可。另外,还是考虑到上述的机制时,优选在回合对象液混合后,经过一定程度的时间静置混合对象液,确保微囊均匀再生的时间。这样的静置时间没有限制,例如设为1分钟以上,优选设为3分钟以上。应当指出,物质包封在微囊空隙部中,可以通过荧光相关光谱法(FluorescenceCorrelationSpectroscopy:FCS)的扩散系数(diffusioncoefficient)变化的检出、尺寸排阻色谱的分离、透射电子显微镜的直接观察等的方法进行确认。在荧光相关光谱法的扩散系数的测定的情况下,作为被包封物质使用荧光材料,通过测定荧光材料的扩散系数的变化,可以确认被包封物质在微囊中聚集(即制得物质包封微囊)(B1-3:混合相关的其他条件)一般,在水性介质中调制含有空微囊以及被包封物质的液体(混合对象液),进行上述的混合。水性介质(水性溶剂)的种类没有限制。优选为水,但在不发生对空微囊的结构达到从好的影响到意想不到的影响,妨碍被包封物质向内部导入的范围内,也可以使用在水中混合其他成分的溶剂(例如生理盐水、水性缓冲液、水和水溶性有机溶剂的混合溶剂等)。作为水性缓冲液可以列出10mMHEPES缓冲液等。作为水溶性有机溶剂可以列出甲醇或乙醇等的醇类、丙酮等的酮类、氯仿等的含氯有机溶剂、二甲醚等的醚系有机溶剂、乙酸乙酯等的酯系有机溶剂等。被混液的调制顺序也是任意的,如后所述,一般,由于将空微囊在水性介质中调制,最好向调制后的空微囊含有液加入被包封物质,进行混合。被包封物质可以直接加入到空微囊含有液中,也可以以水性溶剂中的溶液或悬浊液的形态加入。被混合液中的空微囊以及被包封物质的各自浓度也没有特别的限制,考虑空微囊的结构、被包封物质的种类、与空微囊对应的被包封物质的所期望的包封率等条件决定即可。但是,从提高向空微囊的被包封物质的包封效率的观点来看,将对应水性介质的空微囊的浓度优选设为一般0.1mg/mL以上,中等1mg/mL以上,另外,一般100mg/mL以下,中等10mg/mL以下。特别地,当空微囊的浓度过低时,会有物质包封微囊未形成的情况。应当指出的是,由于可以想到制得的物质包封微囊的粒径依赖于空微囊的浓度,因此空微囊的浓度应该根据所期望的物质包封微囊的粒径而决定。此外,与水性介质对应的被包封物质的浓度,根据被包封物质的性质而不同,但优选设为一般0.1mg/mL以上,中等1mg/mL以上,另外,一般100mg/mL以下,中等10mg/mL以下。特别地,当空微囊的浓度过低时,会有物质包封微囊未形成的情况。被混合液的pH也没有特别的限制,可以考虑空微囊的结构、被包封物质的种类、被混合液中的空微囊以及被包封物质的各自浓度等条件进行适当地调整,但优选为pH5以上,更优选为pH6.5以上,另外,优选为pH9以下,更优选为pH7.5以下。pH的调制通过使用作为溶剂的缓冲液,能够容易地进行。调整被混合液pH使用,有利于保持空微囊的结构并有效地将被包封物质包封在空微囊中。被混合液的离子强度,能够在不破坏空微囊的结构、阻碍被包封物质包封在空微囊中的范围内进行适当调整,但优选为0mM以上,更优选为10mM以上,另外,优选为200mM以下,更优选为50mM以下。被混合液的混合时的温度,在不发生破坏空微囊的结构、阻碍被包封物质包封在空微囊中的范围内没有限制,但优选为10℃以上,更优选为20℃以上,另外,优选为80℃以下,更优选为50℃以下。混合后,可以将形成的物质包封微囊立刻应用于所期望的用途,但为了平衡系统,也可以设立静置混合液的时间。静置混合液的时间,因物质包封微囊的形成效率等条件而不同,优选为50小时以下,更优选为30小时以下。但是,如后所述,在不使用交联剂的情况下,由于形成的物质包封微囊的直径具有随时间增加的倾向,也有优选不设立在微囊均匀再生所需时间以上的静置时间的情况。在使用交联剂的情况下,在含有形成的物质包封微囊的混合液中,加入交联剂进行混合即可。交联剂可以直接添加,也可以调制含有交联剂的水性溶液,将其添加。调制交联剂的水性溶液的水性溶剂、pH、温度、离子强度等调制条件,和被混合液相关的前述条件相同。此外,也可以适当添加另外的透析、稀释、浓缩、搅拌等操作。[B2:空微囊](B2-1:空微囊的结构)在后负载法中,使用具有膜和由所述膜包围城的孔隙部的微囊作为空微囊,其中所述膜由具有不带电亲水性嵌段以及第1带电嵌段的嵌段聚合物的第1聚合物,和具有带与前述第1带电嵌段相反电荷的第2带电嵌段的第2聚合物形成。有关空微囊结构的例子,参照图2~4进行说明。应当指出,图2~4都是模型图,本发明并不限定于这些图。图2为微囊1的局部剖视图。如图2所示,微囊1具有膜1a,和通过膜1a包围成的空隙部1b。图3(a)为与本发明一种方式有关的微囊1的膜1a的局部剖面放大图。如图3(a)所示的膜1a具有由外层1ao、中间层1am以及内层1ai形成的三层结构,主要由第1聚合物2和第2聚合物3形成。图3(b)为图3(a)中所示的第1聚合物2以及第2聚合物3的放大图。如图3(b)所示,第1聚合物2为具有不带电亲水性嵌段2a和第1带电嵌段2b的嵌段聚合物,第2聚合物3为带与第1带电嵌段2b相反电荷的第2带电嵌段3形成的聚合物。因此,如图3(a)所示,优选不带电亲水性嵌段2a形成膜1a的外层1ao,第1带电嵌段2b和第2带电嵌段3静电耦合形成中间层1am。而且优选主要由不带电亲水性嵌段2a形成膜1a的内层1ai。图4(a)为与本发明别的方式有关的微囊1的膜1a的局部剖面放大图。图4(a)所示的膜1a也具有由外层1ao、中间层1am以及内层1ai形成的三层结构,主要由第1聚合物2和第2聚合物3’形成。图4(b)为图4(a)中所示的第1聚合物2以及第2聚合物3’的放大图。如图4(b)所示,第1聚合物2为具有不带电亲水性嵌段2a和第1带电嵌段2b的嵌段共聚物,第2聚合物3’为由不带电亲水性嵌段3a和带与第1带电嵌段2b相反电荷的第2带电嵌段3b形成的聚合物。因此,如图4(a)所示,优选不带电亲水性嵌段2a、3a的一方或两方形成膜1a的外层1ao,第1带电嵌段2b和第2带电嵌段3b静电耦合形成中间层1am。而且优选主要由不带电亲水性嵌段2a、3a的一方或两方形成膜1a的内层1ai。虽然没有理论上的束缚,但由第1聚合物2以及第2聚合物3、3’形成微囊1的机制可以如下考虑。即,图3(b)、图4(b)中所示的第1聚合物2以及第2聚合物3、3’在放置在能够发生电荷相互作用的系统中时,自组装,如图3(a)、图4(a)所示,在带有相互相反电荷的第1带电嵌段2b以及第2带电嵌段3、3b通过静电耦合形成中间层1am的同时,在其外侧配置不带电亲水性嵌段2a、3a并形成外层1ao。另外,优选在中间层1am的内侧中也主要配置不带电亲水性嵌段2a、3a并形成内层1ai。,这样形成如图3(a)、图4(a)所示的三层结构的膜1a,其结果,可以认为形成如图2所示的微囊1。应该指出,微囊1的膜1a可以仅由第1聚合物2以及第2聚合物3、3’形成,也可以在保持以上结构梗概的限制下,含有其他的成分。其他成分没有限制,但可以列出例如,交联剂,带电聚合物、带电分子等。有关交联剂在后文中详细叙述。此外,如后所述微囊1一般在水性介质中调制,此外,由于膜1a的内层1ai主要由不带电亲水性嵌段2a、3a构成,在微囊1的空隙部1b通常存在水性介质(因此本说明书中有将空隙部1b表示为“内水相”的情况)。但是,空隙部1b中也可以存在其他的物质。微囊1的形状没有限制,通常为球形或大致球形。微囊1的粒径根据第1聚合物2以及第2聚合物3、3’的种类及量比、交联剂的有无、微囊1的周边环境(水性介质的种类)等而不同,但优选为10nm以上,更优选为50nm以上,另外,优选为1000nm以下,更优选为400nm以下。微囊1的膜1a的膜的厚度根据第1聚合物2以及第2聚合物3、3’的种类及量比、交联剂的有无、微囊1的周边环境(水性介质的种类)等而不同,但优选为5nm以上,更优选为10nm以上,另外,优选为30nm以下,更优选为15nm以下。(B2-2:第1以及第2聚合物)后负载法中使用的空微囊具有由第1聚合物以及第2聚合物构成的膜。第1聚合物为具有不带电亲水性嵌段和第1带电嵌段的嵌段共聚物。第1聚合物可以仅为1个种类,也可以并用2种以上任意组合及比例。第2聚合物为具有带与第1带电嵌段相反电荷的第2带电嵌段的聚合物。可以是仅由第2带电嵌段形成的聚合物,也可以是具有除第2带电嵌段以外不带电亲水性嵌段的嵌段共聚物。第2聚合物可以是仅为1个种类,也可以并用2种以上任意组合和比例。在2种以上的情况下,也可以并用仅由第2带电嵌段形成的第2聚合物,和具有除了第2带电嵌段以外不带电亲水性嵌段的第2聚合物。第1聚合物以及第2聚合物,除了各种前述的嵌段以外,也可以具有其他嵌段。(B2-2a:不带电亲水性嵌段)第1聚合物具有不带电亲水性嵌段。另外,第2聚合物也可以具有不带电亲水性嵌段。不带电亲水性嵌段为具有不带电且亲水性的性质的聚合物嵌段。这里,“不带电”是指,嵌段整体上为中性。作为例子,可以列出嵌段没有正负电荷的情况。另外,即使在嵌段在分子内具有正负电荷的情况下、局部的实际电荷密度高、如果嵌段整体上电荷被中和到不妨碍通过自组装的形成微囊的程度,也还当作“不带电”。另外,“亲水性”是指针对水性介质表现出溶解性。不带电亲水性嵌段的种类没有限制。可以是单一的重复单元形成的嵌段,也可以是含有任意组合及比例两种以上的重复单元的嵌段。作为不带电亲水性嵌段的具体例子可以列出,聚亚烷基二醇、聚(2-恶唑啉)、多糖、聚乙烯醇、聚乙烯吡咯烷酮、聚丙烯酰胺、聚甲基丙烯酰胺、聚丙烯酸酯、聚甲基丙烯酸酯、聚(2-甲基丙烯酰氧乙基磷酸胆碱)、等电点为在7附近的多肽、蛋白质及它们的衍生物等。其中,优选聚亚烷基二醇、聚(2-恶唑啉)、特别优选聚亚烷基二醇。作为聚亚烷基二醇可以列出聚乙二醇、聚丙二醇等,优选聚乙二醇。不带电亲水性嵌段的分子量没有限制,从促进第1聚合物和第2聚合物的自组装化、有效地制备均质的微囊的观点出发,优选具有规定的范围内的分子量。具体的分子量范围根据不带电亲水性嵌段的种类、和带电嵌段的组合等而不同,但在使用聚乙二醇作为不带电亲水性嵌段的情况下,其分子量(Mw)优选为500以上,更优选为1000以上,另外,优选为15000以下,更优选为5000以下的范围。不带电亲水性嵌段的重复单元数也没有限制,但一般,不带电亲水性嵌段的分子量以满足所述的分子量范围的方式,根据其重复单元的种类而决定。通过使用满足所述条件的不带电亲水性嵌段,能够防止第1聚合物及第2聚合物的水溶液中的缔合、沉淀而稳定化,能够有效地构建微囊。(B2-2b:带电嵌段)第1聚合物具有的第1带电嵌段第2聚合物具有的第2带电嵌段为带有相反电荷的带电嵌段。即,若第1带电嵌段为阳离子性嵌段,则第2带电嵌段为阴离子性嵌段,若第1带电嵌段为阴离子性嵌段,则第2带电嵌段为阳离子性嵌段。(B2-2b-1:阳离子性嵌段)阳离子性嵌段为具有阳离子基团,表现出阳离子性(阳离子性)的聚合物嵌段。但是,阳离子性嵌段在不妨碍第1聚合物和第2聚合物的通过自组装的微囊的形成的范围内,也可以具有一些阴离子基团。阳离子性嵌段的种类也没有限制。可以是由单一重复单元形成的嵌段,也可以是含有任意组合及比例两种以上的重复单元的嵌段。作为阳离子性嵌段,优选多胺,特别优选侧链上具有氨基的聚氨基酸或其衍生物。作为侧链上具有氨基的聚氨基酸或其衍生物,可以列出,聚天冬氨酸、聚谷氨酸、聚赖氨酸、聚精氨酸、聚组氨酸、以及它们的衍生物等,特别地优选聚天冬氨酸衍生物及聚谷氨酸衍生物。阳离子性嵌段的分子量也没有限制,从促进第1聚合物和第2聚合物自组装化、有效地制备均质的微囊的观点出发,优选具有规定的范围内的分子量。阳离子性嵌段的重复单元数也没有限制,一般,以阳离子性嵌段的分子量满足规定范围的方式,根据其重复单元的种类而决定。具体而言,在使用聚天冬氨酸衍生物为阳离子性嵌段的情况下,其重复单元数优选为10以上,更优选为50以上,另外,优选为200以下,更优选为100以下的范围。通过使用满足所述条件的阳离子性嵌段,能够防止第1聚合物及第2聚合物的水溶液中的缔合、沉淀而稳定化,能够有效地构建微囊。(B2-2b-2:阴离子性嵌段)阴离子性嵌段为具有阴离子基团,表现出阴离子性(阴离子性)的聚合物嵌段。但是,阴离子性嵌段在不妨碍第1聚合物和第2聚合物通过自组装的微囊的形成的范围内,也可以具有一些阳离子基团。阴离子性嵌段的种类也没有限制。可以是由单一重复单元形成的嵌段,也可以是含有任意组合及比例两种以上的重复单元的嵌段。作为阴离子性嵌段,优选聚羧酸、聚磺酸、多聚磷酸(核酸)等、特别优选侧链上具有羧基的聚氨基酸或其衍生物、核酸。作为侧链上具有羧基的聚氨基酸或其衍生物可以列出,聚天冬氨酸、聚谷氨酸、向所述的聚阳离子中侧链上具有氨基的聚氨基酸或其衍生物的氨基使用适当量的乌头酸酐和柠康酸酐制得的聚羧酸、以及它们的衍生物等,特别优选聚天冬氨酸、聚谷氨酸。作为核酸,可以列出单链或双链的DNA或RNA。核酸可以是基于微囊用途的功能性核酸。作为功能性核酸可以列出,siRNA、miRNA(微型RNA)、反义RNA、反义DNA、核酶、DNA酶等。它们基于微囊的用途进行选择。例如,在将微囊用作RNAi用的DDS的情况下,使用siRNA作为核酸。另外,核酸可以是进行修饰过的核算。作为修饰后核酸的例子,可以列出,面向微囊稳定化等用途,结合胆固醇或维生素E等疏水性基团的核酸。阴离子性嵌段的分子量没有限制,从促进第1聚合物和第2聚合物自组装化、有效地制备均质的微囊的观点出发,优选具有规定的范围内的分子量。阴离子性嵌段的重复单元数也没有限制,一般,以阴离子性嵌段的分子量满足规定的范围的方式,根据其重复单元的种类而决定。具体而言,在使用聚羧酸、聚磺酸、或核酸作为阴离子性嵌段的情况下,其重复单元数优选为10以上,更优选为50以上,另外,优选为200以下,更优选为100以下的范围。通过使用满足所述条件的阴离子性嵌段,能够防止第1聚合物及第2聚合物的水溶液中的缔合、沉淀而稳定化,能够有效地构建微囊。(B2-2c:不带电亲水性嵌段和带电嵌段的组合)第1聚合物具有不带电亲水性嵌段和第1带电嵌段的组合,另外,在第2聚合物具有除了第2带电嵌段以外不带电亲水性嵌段的情况下不带电亲水性嵌段和第2带电嵌段的组合,都没有限制,可以为任意的不带电亲水性嵌段和任意的带电嵌段能够组合(应该指出,下面的记载中,有第1带电嵌段以及第2带电嵌段一起表示为“带电嵌段”的场合)。不带电亲水性嵌段以及带电嵌段的个数也是任意的,各自可以为一个也可以为两个以上,在两个以上的情况下,可以相互相同,也可以不同。不带电亲水性嵌段以及带电嵌段的结合方式也没有限制,可以直接结合,也可以通过连接基团结合。作为连接基的例子,可以列出具有与不带电亲水性嵌段以及带电嵌段的总个数对应个数的烃基。作为连接基团的烃基可以是脂肪族可以是芳香族也可以是连接它们的物质,在脂肪族的情况下,可以是不饱和也可以是饱和,另外,直链、支链、环状都可以。作为连接基团的烃基的分子量没有限制,一般为5000以下,优选为1000以下。作为连接基团的烃基的例子,可以列出,没食子酸衍生物、3,5-二羟基苯甲酸衍生物、甘油衍生物、环己烷衍生物、L-赖氨酸等,优选为3,5-二羟基苯甲酸衍生物等。作为连接基团的其他例子,可以列出二硫基。二硫基用于连接一个不带电亲水性嵌段和一个带电嵌段。通过二硫基将不带电亲水性嵌段和带电嵌段连接,由于放置微囊的环境或外部的作用使二硫基开裂,能够使微囊的形态或性质发生变化。利用这一点,例如在微囊中包封药物,制得的物质包封微囊用于药物输送用的DDS的情况下,可以考虑通过使生物体内二硫基的开裂,能够促使微囊内包封的物质的释放。此外,第1带电嵌段和第2带电嵌段的比率(阳离子性嵌段和阴离子性嵌段的比率)、以及,不带电亲水性嵌段和带电嵌段的比率也是任意的,从促进第1聚合物和第2聚合物自组装化、有效地制备均质的微囊的观点出发,优选在以下基准中选择。首先,有关阳离子性嵌段和阴离子性嵌段的比率,理想地,调整下式(i)中规定的C/A比,通常为0.3,优选为0.5以上,更优选为0.6以上,另外,一般小于3.0,优选为2.0以下,更优选为1.7以下。[数学式1]这里,第1以及第2聚合物中阳离子基团以及阴离子基团的摩尔数为与阳离子性嵌段以及阴离子性嵌段的结构有关的值,能够通过普通的电位差(酸、盐)滴定获得。此外,第1以及第2聚合物中不带电亲水性嵌段和带电嵌段的比率,优选考虑满足所述C/A比的范围的阳离子性嵌段以及阴离子性嵌段的比率而决定。具体而言,将下式(ii)中规定的分子量比率X,理想地,在通常为0.01以上,优选为为0.05以上,另外,通常为0.35以下,优选为为0.1以下的范围内获取。在使用每一种阳离子性嵌段(设为一个单体具有一个正电荷)以及阴离子性嵌段(设为一个单体具有一个负电荷),将不带电亲水性嵌段导入到至少一方中而使用的情况下(即,在第1聚合物为由阳离子性或阴离子性嵌段和不带电亲水性嵌段形成的嵌段聚合物,第2聚合物为由阴离子性或阳离子性嵌段的单独聚合物,或是其与不带电亲水性嵌段形成的嵌段聚合物的情况下),其X通过下式规定。[数学式2]MNA表示连接阴离子性嵌段的不带电亲水性嵌段的分子量,MNC表示连接阳离子性嵌段的不带电亲水性嵌段的分子量,MC表示阳离子性嵌段的分子量,MA表示阴离子性嵌段的分子量,PC表示阳离子性嵌段的聚合度,PA表示阴离子性嵌段的聚合度。(B2-2d:第1以及第2聚合物的具体例)作为第1及第2聚合物的具体例,可以列出以下的“例1”,“例2”。[例1]作为第1聚合物使用下记(A1),作为第2聚合物使用下记(B1)。(A1)含有不带电亲水性嵌段和阴离子性嵌段的嵌段共聚物。(B1)下记(i)的嵌段共聚物和/或下记(ii)的聚合物。(i)含有不带电亲水性嵌段和阳离子性嵌段的嵌段共聚物。(ii)含有阳离子性嵌段的聚合物(但是不含不带电亲水性嵌段)。[例2]作为第1聚合物使用下记(A2),作为第2聚合物使用下记(B2)。(A2)含有不带电亲水性嵌段和阳离子性嵌段的嵌段共聚物。(B2)下记(iii)的嵌段共聚物和/或下记(iv)的聚合物。(iii)含有不带电亲水性嵌段和阴离子性嵌段的嵌段共聚物。(iv)含有阴离子性嵌段的聚合物(但是不含不带电亲水性嵌段)。应该指出,在本发明中,如所述(B1)(ii)以及(B2)(iv)的聚合物那样,为方便起见有将不含不带电亲水性嵌段的聚合物称为均聚物的情况。作为所述(B1)(i)、(ii)以及(A2)的各聚合物的阳离子性嵌段,没有限制,可以列出,例如优选为侧链上具有阳离子基团的由多肽衍来的物质。同样地,所述(A1)以及(B2)(iii)、(iv)的各聚合物中,作为阴离子性嵌段,没有限制,可以列出,例如优选为由侧链上具有阴离子基团的多肽或核酸来的物质。更具体而言,作为所述(A1)以及(B2)(iii)的各嵌段聚合物,可以列出例如优选下述通式(I)和/或(II)表示的物质。[化学式I]这里,通式(I)以及(II)的结构式中,重复单元数(聚合度)“m”的嵌段为来自PEG的不带电亲水性嵌段(下面有表示为“PEG嵌段”的情况),重复单元数“n-y”的部分和“y”的部分合起来的嵌段为来自聚阴离子的阴离子性嵌段(下面有表示为“聚阴离子嵌段”的情况)。通式(I)以及(II)中R1a以及R1b表示各自独立的氢原子或未取代或取代了的直链或支链的C1-12的烷基。作为直链或支链的C1-12例如,可以列出,甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、正戊基、正己基、癸基、十一烷基等。另外,作为取代了的情况的取代基可以列出缩醛化甲酰基、氰基、甲酰基、羧基、氨基、C1-6烷氧羰基、C2-7酰胺基,相同或不同的3-C1-6烷基甲硅烷氧基,甲硅烷氧基,或甲硅烷氨基等。其中,缩醛化意味着通过甲酰基的羰基和例如碳原子数1~6的链烷醇的2分子或碳原子数2~6的支链即可的的亚烷基二醇反应形成的缩醛部分,也是该羰基的保护方法。例如,当取代基为缩醛化甲酰基的时,在酸性温和的条件下加水分解,可以转化为作为其他取代基的甲酰基(-CHO)(或醛基)。通式(I)以及(II)中L1以及L2表示连接基团。具体而言,L1优选为-(CH2)b-NH-(其中b为1~5的整数),L2优选为为-(CH2)c-CO-(其中c为1~5的整数)。通式(I)以及(II)中R2a、R2b、R2c以及R2d各自独立地表示亚甲基或亚乙基。R2a以及R2b都是亚甲基的情况相当于聚(天冬氨酸衍生物),都是亚乙基的情况相当聚(谷氨酸衍生物),另外,R2c以及R2d都是亚甲基的情况相当聚(天冬氨酸衍生物),都是亚乙基的情况相当聚(谷氨酸衍生物)。这些通式中,在R2a以及R2b(R2b以及R2a)表示亚甲基以及亚乙基两者的情况下,以及在R2c以及R2d(R2d以及R2c)表示亚甲基以及亚乙基两者的情况下,天冬氨酸衍生物和谷氨酸衍生物的重复单位,各自形成嵌段存在,或是能够随机存在。通式(I)以及(II)中R3表示氢原子、保护基团、疏水性基团或是聚合性基团。具体而言,R3优选为乙酰基,丙烯酰基或甲基丙烯酰基。通式(I)以及(II)中R4表示羟基、氧代苄基、-NH-(CH2)a-X基团或引发剂残基。其中,a为1~5的整数,X优选为含有伯、仲、叔胺或季铵盐中的一种或两种以上的胺类化合物残基,或是,没有胺的化合物残基。而且,根据情况,R4优选为-NH-R9(其中,R9表示未取代或取代了的直链或支链的C1-20烷基)。通式(I)以及(II)中,m为5~2000的整数,优选为2~270的整数,更优选为10~100的整数。另外,n为2~5000的整数,y为0~5000的整数,n以及y优选为5~300的整数,更优选为10~100的整数。但是,y小于n。通式(I)以及(II)中的各重复单元,以记载的方便起见按特定顺序表示,但各重复单元可以按随机的顺序存在。特别是,仅在有关聚阴离子嵌段中的各重复单元,优选以如上所述的随机顺序存在获得。通式(I)以及(II)表示的嵌段共聚物的分子量(Mw)没有限制,优选为3000~30000,更优选为5000~20000。另外,关于各个嵌段,PEG嵌段的分子量(Mw)优选为500~5000,更优选为1000~5000,聚阴离子嵌段的分子量(Mw)整体优选为500~50000,更优选为1000~20000。通式(I)以及(II)所表示的嵌段聚合物的制备方法没有限制,例如,可以列出,预先合成含有R1aO–或R1bO–和PEG链嵌段部分的嵌段(PEG嵌段),在该PEG嵌段的段末端(R1aO–或R1bO–相反的末端),选定的单体按顺序聚合,其后,根据需要以在侧链含有阴离子性基团的方式进行取代或变换的方法,或者,预先合成上述PEG嵌段和具有含阴离子性基团侧链的嵌段部分,它们相互结合的方法。该制备方法的各种反应的方法及条件可以参考常规方法适当选择或设定。上述PEG嵌段,例如,可以使用WO96/32434号公报、WO96/33233号公报以及WO97/06202号公报等记载的嵌段共聚物的PEG嵌段部分的制备方法进行调制。作为通式(I)以及(II)表示的嵌段聚合物的更具体的制备方法,可以列出,例如,优选使用在末端具有氨基的PEG嵌段衍生物,在其氨基的末端,聚合β-苄基-L-天冬氨酸(BLA)以及Nε-Z-L-赖氨酸等保护氨基酸的N-羧酸酐(NCA)合成嵌段聚合物,其后,以各嵌段的侧链成为具有所述阴离子性基团的侧链的方式,进行取代或变换的方法本发明中,作为通式(I)以及(II)表示的嵌段聚合物的具体例子,,可以列出,例如,优选由不带电亲水性嵌段的聚乙二醇(下面有表示为“PEG”的情况)和,阴离子性嵌段的聚天冬氨酸(下面有表示为“P(Asp)”的情况)形成的下式的阴离子性嵌段聚合物(下面有表示为“PEG-P(Asp)”的情况)等。(应该指出,以下的式子中作为平衡阳离子的例子有表示为Na+的情况,但是平衡阳离子不限制于此)。[化学式2]上式中,m为表示PEG聚合度的整数。n为表示P(Asp)聚合度的整数。a、b都为大于0小于1的数。但是a+b=1。作为PEG-P(Asp),PEG嵌段的分子量(Mw)优选:2000、表示聚阴离子嵌段的P(Asp)的单元数(上式中n)优选:70或75。作为所述(A2)以及(B1)(i)的各嵌段聚合物,可以列出,例如优选下述通式(III)和/或(IV)表示的物质。[化学式3]这里,通式(III)以及(IV)的结构式中,重复单元数(聚合度)“m”的嵌段为来自PEG的不带电亲水性嵌段(PEG嵌段),重复单元数“n-y-z”的部分和“y”部分和“z”的部分合起来的嵌段为来自聚阳离子的阳离子性嵌段(以下,聚阳离子嵌段)。通式(III)以及(IV)中R1a以及R1b表示各自独立的氢原子或未取代或取代了的直链或支链的C1-12的烷基。作为直链或支链的C1-12例如,可以列出,甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、正戊基、正己基、癸基、十一烷基等。另外,作为取代了的情况的取代基可以列出,缩醛化甲酰基、氰基、甲酰基、羧基、氨基、C1-6烷氧羰基、C2-7酰胺基,相同或不同的3-C1-6烷基甲硅烷氧基,甲硅烷氧基,或甲硅烷氨基等。其中,缩醛化意味着通过甲酰基的羰基和,例如碳原子数1~6的链烷醇的2分子或碳原子数2~6的支链即可的亚烷基二醇反应形成的缩醛部分,也是该羰基的保护方法。例如,当取代基为缩醛化甲酰基的时,在酸性温和的条件下加水分解,可以转化为其他取代基的甲酰基(-CHO:或醛基)。通式(III)以及(IV)中L1以及L2表示连接基团。具体而言,L1优选为-(CH2)b-NH-(其中b为1~5的整数),L2优选为-(CH2)c-CO-(其中c为1~5的整数)。通式(III)以及(IV)中R2a、R2b、R2c以及R2d各自独立地表示亚甲基或亚乙基。R2a以及R2b都是亚甲基的情况相当聚(天冬氨酸衍生物),都是亚乙基的情况相当聚(谷氨酸衍生物),另外,R2c以及R2d都是亚甲基的情况相当聚(天冬氨酸衍生物),都是亚乙基的情况相当聚(谷氨酸衍生物)。这些通式中,在R2a以及R2b(R2b以及R2a)表示亚甲基以及亚乙基两者的情况下,以及在R2c以及R2d(R2d以及R2c)表示亚甲基以及亚乙基两者的情况下,天冬氨酸衍生物和谷氨酸衍生物的重复单位,各自形成嵌段存在,或是能够随机存在。通式(III)以及(IV)中R3表示氢原子、保护基团、疏水性基团或是聚合性基团。具体而言,R3优选为乙酰基,丙烯酰基或甲基丙烯酰基。通式(III)以及(IV)中R4表示羟基、氧代苄基、-NH-(CH2)a-X基团或引发剂残基。其中,a为1~5的整数,X优选为含有伯、仲、叔胺、季铵盐中的一种或两种以上的胺类化合物残基,或是,没有胺的化合物残基。而且,根据情况,R4优选为-NH-R9(其中,R9表示未取代或取代了的直链或支链的C1-20烷基)。通式(III)以及(IV)中R5a、R5b、R5c以及R5d各自独立地表示羟基、氧代苄基、-NH-(CH2)a-X基团或引发剂残基。其中,a为1~5的整数,X优选为含有伯、仲、叔胺、季铵盐中的一种或两种以上的胺类化合物残基,或是,没有胺的化合物残基。R5a和R5b的总数以及R5c和R5d的总数中,作为-NH-(CH2)a-X基团(其中,X为(NH(CH2)2)e-NH2(但是e为0~5的整数))的物质,优选至少存在两个以上,更优选存在上述总数的50%以上,进一步优选存在上述总数的85%以上。另外,优选,R5a、R5b、R5c以及R5d的全部或一部分优选为-NH-(CH2)a-X基团(其中,a为2,X为(NH(CH2)2)e-NH2(但是e为1))。更进一步地,作为R4和R5a、R5b、R5c以及R5d的示例,上述-NH-(CH2)a-X基团中,X特别优选从下述的各式所表示的基团中选择的情况。[化学式4]其中,上述各式中,X2表示氢原子或C1-6烷基或氨基C1-6烷基,R7a、R7b以及R7c各自独立地表示氢原子或甲基,d1、d2以及d3各自独立地表示1~5的整数,e1、e2以及e3各自独立地表示1~5的整数,f表示0~15的整数,g表示0~15的整数,R8a以及R8b各自独立地表示氢原子或保护基。其中,该保护基,优选选自由通常用作氨基保护基的Z基、Boc基、乙酰基以及三氟乙酰基组成的组中的基团。通式(III)以及(IV)中R6a以及R6b各自独立地为氢原子、-C(=NH)NH2、或保护基,其中保护基优选选自由通常用作氨基保护基的Z基、Boc基、乙酰基以及三氟乙酰基组成的组中的基团。另外,通式(III)以及(IV)中,t优选为2~6的整数,更优选为3或4。通式(III)以及(IV)中,m为5~2000的整数,优选为2~270的整数,更优选为10~100的整数。另外,n为2~5000的整数,y为0~5000的整数,z为0~5000的整数。N优选为5~300的整数,更优选为0或10~100的整数。y以及z优选为0或5~300的整数,更优选为0或10~100的整数。但是,y和z的和(y+z)不大于n。通式(III)以及(IV)中个重复单元,记载的方便起见按特定顺序表示,但各重复单元可以按随机的顺序存在。特别地,仅在关于聚阳离子嵌段中的各重复单元,优选为存在如上所述以随机顺序而获得物质。通式(III)以及(IV)表示的嵌段聚合物的分子量(Mw)没有限制,优选为23000~45000,更优选为28000~34000。另外,关于各个嵌段,PEG嵌段的分子量(Mw)优选为500~15000,更优选为1000~5000,聚阳离子嵌段的分子量(Mw)整体优选为500~50000,更优选为1000~30000。通式(III)以及(IV)表示的嵌段的制备方法没有限制,例如,可以列出,预先合成含有R1aO–或R1bO–和PEG链嵌段部分的嵌段(PEG嵌段),在该PEG嵌段的段末端(R1aO–或R1bO–相对的末端),选定的单体按顺序聚合,其后,根据需要以侧链含有阳离子性基团的方式进行取代或变换的方法,或者,预先合成上述PEG嵌段和具有含阳离子性基团侧链的嵌段部分,将它们相互结合的方法。有关该制备方法的各种反应的方法及条件可以参考常规方法适当选择或设定。上述PEG嵌段,例如,可以使用WO96/32434号公报、WO96/33233号公报以及WO97/06202号公报等记载的嵌段共聚物的PEG嵌段部分的制备方法进行调制。作为通式(III)以及(IV)表示的嵌段聚合物的更具体的制备方法,可以列出,例如,优选使用末端具有氨基的PEG嵌段衍生物,在其氨基的末端,聚合β-苄基-L-天冬氨酸(BLA)以及Nε-Z-L-赖氨酸等保护氨基酸的N-羧酸酐(NCA)合成嵌段聚合物,其后,以各嵌段的侧链成为具有所述阳离子性基团的侧链方式,用二乙烯三胺(DET)等进行取代或变换的方法作为通式(III)以及(IV)表示的嵌段聚合物的具体例子,,可以列出,例如,优选由不带电亲水性嵌段的聚乙二醇(下面有表示为“PEG”的情况),和阳离子性嵌段的聚(含有二氨基结构的天冬氨酸)(下面有表示为“P(Asp-AP)的情况”)形成的下式的阳离子性嵌段聚合物(下面有表示为“PEG-P(Asp-AP)”的情况)等。(应该指出,以下的式子中作为平衡阴离子的例子有表示为Cl-的情况,但是平衡阴离子不限定于此)。[化学式5]上式中,m为表示PEG聚合度的整数。n为表示P(Asp-AP)聚合度的整数。a、b都为大于0小于1的数。但是a+b=1。作为PEG-P(Asp-AP),特别优选PEG嵌段的分子量(Mw):2000、表示聚阳离子嵌段的P(Asp-AP)的单元数(上式中n):70或75。作为所述(B2)(iv)的聚合物,可以列出,例如优选下述通式(V)和/或(VI)表示的物质。应该指出,有关通式(V)以及(VI)的说明,可以同样适当适用有关所述通式(I)以及(II)的说明(但是关于PEG嵌段的说明除外)。[化学式6]本发明中,作为通式(V)以及(VI)表示的聚合物的具体例子,可以列出,优选例如由阴离子性嵌段的聚天冬氨酸(P(Asp))形成的下式阴离子性均聚物(以下有表示为“Homo-P(Asp)的情况”)。[化学式7]上式中,n为表示P(Asp)聚合度的整数。a、b都为大于0小于1的数。但是a+b=1。特别优选,作为Homo-P(Asp),表示聚阴离子嵌段的P(Asp)的单元数(上式中n):70或82。作为所述(B1)(ii)的聚合物,可以列出,优选例如下述通式(VII)和/或(VIII)表示的物质。应该指出,有关通式(VII)以及(VIII)的说明,可以同样适当适用有关所述通式(III)以及(IV)的说明。[化学式8]本发明中,作为通式(VII)以及(VIII)表示的聚合物的具体例子,可以列出,优选例如由阳离子性嵌段的聚(含有二氨基结构的天冬氨酸)(下面有表示为“P(Asp-AP)的情况”)形成的下式阳离子性均聚物(以下有表示为“Homo-P(Asp)的情况”)。[化学式9]n为表示P(Asp)聚合度的整数。a、b都为大于0小于1的数。但是a+b=1。特别优选,作为Homo-P(Asp),表示聚阴离子嵌段的P(Asp-AP)的单元数(上式中n):70或82。(B2-3:其他的膜成分)在空微囊形成时,除了第1聚合物以及第2聚合物以外,在不妨碍微囊的形成,不降低稳定性的范围内,可以添加其他的膜成分。其他的膜成分没有特别的限制,作为具体例,可以列出,带电聚合物、带电纳米粒子等。作为带电聚合物,可以列出,具有1个或2个以上所述带电嵌段(阳离子性嵌段或阴离子性嵌段)的聚合物,不适用所述第1聚合物以及第2聚合物的任意带电聚合物。作为带电纳米粒子,可以列出,表面具有电荷的金属系纳米粒子等。所述其他的膜成分,可以单独使用一种,也可以任意组合及比例的并用两种以上。所述其他的膜成分使用量也没有限制,优选限制在不妨碍第1聚合物和第2聚合物自组装形成微囊的程度。具体而言,最好通常为微囊总重量的30%以下,优选为20%以下,更优选为10%以下。(B2-4:空微囊的制备方法)包封物质的空微囊,由于是利用第1聚合物和第2聚合物的静电相互作用形成的,因此通过将第1聚合物和第2聚合物在水性介质中混合,可以简便地制备。根据这样的制备方法,由于不使用有机溶剂就能制备得到微囊,因此在DDS或生物材料等领域中是具有优势。具体而言,准备含有第1聚合物形成的第1水性溶液,和含有第2聚合物形成的第2水性溶液。第1以及第2水性溶液,根据要求也可以进行过滤精制。第1水性溶液中第1聚合物的浓度,以及,第2水性溶液中第2聚合物的浓度没有限制,考虑第1聚合物和第2聚合物的总电荷数的比率,第1聚合物以及第2聚合物的水性溶液中的溶解度、微囊的形成效率等条件,适当决定。第1以及第2水性溶液的溶剂,如果是水性溶剂,其种类没有限制。优选为水,在不妨碍微囊形成的范围内,也可以使用在水中混合其他成分的溶剂,例如生理盐水、水性缓冲液、水和水溶性有机溶剂的混合溶剂等。作为水性缓冲液,可以列出10mMHEPES缓冲液等。第1以及第2水性溶液的pH能够在不妨碍微囊形成的范围内适当调整,但优选pH5以上,更优选pH6.5以上,另外,优选pH9以下,更优选pH7.5以下。pH的调整通过使用作为溶剂的缓冲液,可以容易地进行。对于第1以及第2水性溶液的pH调整后使用的操作,有利于保持第1聚合物以及第2聚合物的带电状态,有效地形成微囊。第1以及第2水性溶液的温度,根据第1聚合物以及第2聚合物在溶剂中的溶解度适当决定,但优选为10℃以上,更优选为20℃以上,另外,优选为80℃以下,更优选为50℃以下。第1以及第2水性溶液的离子强度能够在不妨碍微囊形成的范围内适当调整,但优选为0mM以上,更优选为10mM以上,另外,优选为200Mm以下,更优选为50mM以下。通过将以上的第1以及第2水性溶液混合,制备出微囊。混合的方法没有限制,可以向第1水性溶液中加入第2水性溶液,也可以向第2水性溶液加入第1水性溶液。另外,也可以将第1混合溶液以及第2混合溶液同时加入容器中混合。可以将获得的第1及第2水性溶液的混合液适当搅拌。第1以及第2水性溶液混合时的温度,在不妨碍微囊形成的范围内没有限制,优选考虑与第1聚合物以及第2聚合物对应的温度的溶解度进行设定。具体而言,优选为10℃以上,更优选20℃以上,另外,优选为60℃以下,更优选为50℃以下。混合后,可以将形成的空微囊立刻提供给后负载法,为了平衡体系,也可以设置静置混合液的时间。但是,由于形成的微囊的直径有随时间增大的倾向,因此通常不设置静置时间,优选将形成的微囊立刻提供给后负载法。在使用其他的膜成分的情况下,将上述的第1以及第2水性溶液,和该膜成分混合即可。此时,混合前的第1或第2水性溶液中可以加入该膜成分混合,最好在该膜成分和第1或第2水性溶液之间,没有妨碍微囊形成的聚合或相互作用。另外,可以在第1及第2水性溶液混合时将该膜成分一起加入混合,也可以在第1以及第2水性溶液混合后加入该膜成分进一步混合。另外,可以将其他的膜成分直接混合使用,也可以调制含有该膜成分的水性溶液,使用其混合。调制膜成分的水性溶液中的水性溶剂、pH、温度、离子强度等调制条件与第1以及第2水性溶液相关的上述条件相同。另外,也可以进一步地适当添加透析、稀释、浓缩、搅拌等操作。(B3:被包封物质)微囊内包封的被包封物质没有限制,可以根据物质包封微囊的用途或性质等,适当、任意地选择。特别地,按照现有制备方法的一种同时混合法,在使用带电物质作为被包封物质的情况下,作为膜构成成分的聚合物的自组装的微囊的形成由于被包封物质的电荷而被阻碍,具有不能获得合适的物质包封微囊的问题。但是,根据后负载法,对被包封物质的电学性质没有这样的限制,即使是在使用带电物质、不带电物质作为被包封物质的情况下,都能够有效地形成物质包封微囊。具体而言,作为被包封物质的种类,可以列出生物分子、有机化合物、无机物质等。作为生物分子,可以列出蛋白质、多肽、氨基酸、核酸(DNA、RNA)、脂质(脂肪酸、甘油酯、类固醇等)、糖类(单糖、多糖)、以及它们的衍生物、和它们的两种以上结合的物质等(糖蛋白、糖脂等)。其中,优选为蛋白质、糖等。作为有机化合物,可以列出,发光(荧光、磷光等)分子、水溶性药剂、水溶性高分子、平均粒径100nm以下的水溶性分子聚合体(胶束、微囊、纳米凝胶等)、平均粒径100nm以下的乳液等。其中优选为平均粒径50nm以下的高分子胶束、分子量10万以下的水溶性高分子。作为无机物质,可以列出,能够在水中分散的金属纳米粒子、氧化物纳米粒子(二氧化硅纳米颗粒,二氧化钛纳米颗粒,氧化铁纳米颗粒等)、半导体纳米粒子(量子点等)、水溶性碳簇、硼簇、金属络合物等。其中,优选为平均粒径20nm以下的量子点。另外,按照用途分类,作为被包封物质,可以列出,抗癌剂(例如,多柔比星或紫杉醇等疏水性抗癌剂、顺铂等金属络合物抗癌剂等及它们的高分子胶束体)、MRI等诊断使用的钆或铁化合物、有机发光(荧光、磷光等)色素或量子体等。被包封物质的分子量以及粒径也没有限制,从被包封物质有效地包封在空微囊中的观点出发,被包封物质的分子量通常为20万以下,其中优选为10万以下,被包封物质的粒径通常为100nm以下,其中优选为50nm以下。相对空微囊的被包封物质的使用比率也可以在不发生破坏空微囊结构、阻碍被包封物质向空微囊内包封的范围内,根据所期望的被包封物质的包封量进行调整。被包封物质可以单独使用1种,也可以使用2种以上的任意比率及组合。[B4:其他的工序]后负载法,至少具有准备有指定结构的空微囊的工序和将所述空微囊以及被包封物质在水性介质中混合的工序即可,还可以进一步具有其他的工序。可以列出,例如,交联剂处理、过滤操作、透析操作、冷冻干燥操作等。其中,在将物质包封微囊在生理环境下或生理盐水中等的盐存在的条件下使用的情况(例如作为DDS使用的情况)中,从防止粒径的随时间增大的观点来看,,对形成的物质包封微囊,优选实施作为后处理的交联剂处理。即,在生理环境下或生理盐水中等的盐存在的条件下,虽然具有不含交联剂的微囊的粒径有随时间增大的倾向,但通过实施交联剂处理,可以防止粒径增大。交联剂的种类没有限制,可以根据微囊的用途或第1聚合物以及第2聚合物的种类、其他膜成分的种类等适当选择,但从有效地进行交联,和提高物质包封微囊稳定性的观点出发,优选为与第1聚合物以及第2聚合物的带电嵌段具有的带电基团(例如,氨基等阳离子基团或羧基等阴离子基团)反应,和被包封物质不反应的交联剂。作为交联剂的具体例子,可以列出,交联氨基的交联剂(例如,戊二醛、二甲基辛二硝酸二盐酸脂(dimethylsuberimidatedihydrochloride:DMS)、3,3’-二硫基二亚胺代丙酸二甲酯(dimethyl3,3'-dithiobispropionimidate:DTBP))、交联氨基和羧基缩合的交联剂(例如,1-乙基-3-(3-二甲氨基丙基)碳化二亚胺(1-ethyl-3-(3-dimethylaminopropy)carbodiimide:EDC)等)、交联磷酸基团的交联剂(例如,钙离子等金属离子)等,优选戊二醛、EDC等,特别优选EDC。另外。可以单独使用一种交联剂,也可以使用两种以上的任意组合及比率。交联剂的使用量也没有限制,考虑交联剂的种类以及交联点的数量、交联对象成分的量等,适当决定即可,例如,在交联氨基和羧基的交联剂的情况下,最好以下式(iii)中规定的CL比满足随后说明条件的方式选择使用量。[数学式3]从有效地进行交联,和提高物质包封微囊稳定性的观点出发,CL比通常为0.1以上,优选为0.5以上,最好调整交联剂和第1以及第2聚合物的量比。另一方面,在使用物质包封微囊作为DDS输送药物的情况下(例如使用的情况),从使药物在靶位高效地释放的观点出发,交联剂的使用量优选不能过多,具体而言,CL比通常为10以下,优选为5以下,最好调整交联剂和第1以及第2聚合物的量比。但是,上述记载的CL比的范围仅是指导,实际上最好根据微囊的用途和第1聚合物以及第2聚合物的种类、其他膜成分的种类等,适当调制CL比。[B5:物质包封微囊]通过以包括后负载法的各种任意的方法将物质包封在空微囊中,所述空微囊的空隙部内包封有所述的被包封物质,制得物质包封微囊。这样的物质包封微囊含有膜、所述膜包围成的孔隙部、在所述孔隙部内包封的物质,其中所述膜是由具有作为不带电亲水性嵌段及第1带带你嵌段的嵌段共聚物的第1聚合物和具有带与所述第1带电嵌段相反电荷的第2带电嵌段的第2聚合物形成的。物质包封微囊的膜的结构,和上述空微囊的膜的结构基本相同。即,物质包封微囊优选具有和用图2~4说明的空微囊的结构膜相同的三层结构膜,其形状通常为球形或大致球形。后负载法制得的物质包封微囊的粒径,根据空微囊的结构、被包封物质的种类、微囊周围环境(水性介质的种类)、混合的条件等而不同,但通常和空微囊的粒径大致相同。另外,不论后负载法或其他方法,制得的物质包封微囊的粒径,优选为10nm以上,更优选为50nm以上,另外,优选为1000nm以下,更优选为400nm以下,最优选为200nm以下。后述的本法的各物质包封微囊的粒径,除非另有说明,也是相同程度。后负载法制得的物质包封微囊的膜厚度也根据空微囊的结构、被包封物质的种类、微囊周围环境(水性介质的种类)、混合的条件等而不同,但通常和空微囊的膜厚度大致相同。另外,不论后负载法或其他方法,制得的物质包封微囊的膜厚度,优选为5nm以上,更优选为10nm以上,另外,优选为30nm以下,更优选为15nm以下。后述的本法的各物质包封微囊的膜厚度,除非另有说明,也是相同程度。现有的方法中,尽管一部分地能够将物质包封于静电相互作用型微囊(PICsome)中,但适用范围是有限制的。另外,难以从后面的工序中将物质包封到空微囊中,或是不可能的。根据后负载法,能够制备将广泛的物质包封在静电相互作用型微囊(PICsome)中的物质包封静电相互作用型微囊(PICsome)(物质包封PICsome)。物质包封微囊,由于在经由高分子自组装而形成的微囊的空隙部内,稳定地保持各种物质,因此能有效地在传送药物的DDS,和负载有效成分的功能性材料等的各种用途中使用。例如,通过使用siRNA(smallinterferingRNA)作为被包封物质或微囊膜的构成成分,可以将获得的物质包封微囊用于RNAi(RNA干涉)用的DDS等。另外,通过包封多种的药剂作为包封物质,或作为被包封物质以及膜材料并用,在药剂的联合疗法也能使用。[C]物质包封交联微囊及其制备方法<C-1:第1形态/物质包封交联微囊的单分散体及其制备方法>[概要]第1方式的方式为物质包封交联微囊的制造方法,包括将空的交联微囊的单分散体在同时含有目标物质和水性介质的混合液中混合,形成在内水相中包封有目标物质的物质包封交联微囊的单分散体的工序。第1方式的方法中,混合液中含有的目标物质的浓度,在非交联的方面将与空的交联微囊的单分散体不同的空的非交联微囊的单分散体在同混合液中混合的情况下,阻碍在内水相中包封目标物质的物质包封非交联微囊的单分散体形成的浓度。在将空的非交联微囊的单分散体,在目标物质存在下,水性介质中混合的情况下,当目标物质的浓度超过一定浓度时,就会有阻碍物质包封非交联微囊的单分散体的形成的(即,形成多分散聚合物)情况。对此,在将空的交联微囊的单分散体,在目标物质存在下,在水性介质中混合的情况下,即使超过该一定浓度,也不阻碍物质包封交联微囊的单分散体的形成。而且,根据第1方式的方法,水性介质中存在的目标物质的浓度,即使与传统方法相比为高浓度(例如,后负载法中阻碍物质包封微囊的单分散体形成的浓度),也可以在继续保持其单分散性的同时,将目标物质封入空的交联微囊中,据此,可以制备与现有方法相比包封有高浓度的目标物质的物质包封微囊的单分散体。[空的交联微囊的单分散体]空的交联微囊的单分散体的平均粒径,通常为30nm以上,优选为50nm以上,更优选为70nm以上,通常为10000nm以下,优选为1000nm以下,更优选为400nm以下。构成空的交联微囊的单分散体的各微囊为含有第1以及第2聚合物的,包括第1和/或第2聚合物交联成的交联膜和由交联膜包围成的内水相的,在内水相中不包含目标物质的微囊。空的交联微囊中的第1以及第2聚合物如上述[B2-2:第1以及第2聚合物]中记载。空的交联微囊中的其他膜成分如上述[B2-3:其他的膜成分]中记载。空的交联微囊的交联膜中含有的第1和/或第2聚合物进行交联。即,空的交联微囊的交联膜含有选自由第1聚合物间的交联键、第2聚合物间的交联键、以及第1聚合物和第2聚合物间的交联键组成的组中的1种或2种以上的交联键。空的交联微囊的交联膜,从提升形成的物质包封交联微囊的单分散性的观点出发,优选含有在第1聚合物和第2聚合物间形成的交联键。空的交联微囊的交联膜中含有的第1和/或第2聚合物,优选通过选自由阳离子基团间形成的交联键、阴离子基团间形成的交联键、以及阳离子基团和阴离子基团间形成的交联键组成的组中的1种或2种以上的交联键进行交联,更优选为通过阳离子基团和阴离子基团间形成的交联键进行交联。阳离子基团以及阴离子基团为具有第1聚合物(第1带电嵌段)以及第2聚合物(第2带电嵌段)的带电基团。交联率,在目标物质封入时维持空的交联微囊的单分散性的范围内,可以根据目标物质的种类、目标物质封入时使用的混合方法的种类等进行适当调节。其中,“交联率”为在交联膜中含有的阳离子基团和/或阴离子基团的总摩尔数中形成有交联键的比例,即,在阳离子基团间交联的情况下,在交联膜中含有的阳离子基团的总摩尔数中形成有阳离子基团间的交联键的比例,在阴离子基团间交联的情况下,在交联膜中含有的阴离子基团的总摩尔数中形成有阴离子基团间的交联键的比例,在阳离子基团和阴离子基团间交联的情况下,交联膜中含有的阳离子基团以及阴离子基团的总摩尔数中形成有阳离子基团和阴离子基团交联的比例。空的交联微囊的交联率越高,空的交联微囊中能够封入的目标物质的最大分子尺寸(最大分子量)越小,另一方面,空的交联微囊的交联率越低,空的交联微囊中能够封入的目标物质的最大分子尺寸(最大分子量)越大。因此,目标物质的分子尺寸(分子量)越小,交联率的上限越高,另一方面,目标物质的分子尺寸(分子量)越大,交联率的上限越低。从扩大能够封入的目标物质的分子尺寸(分子量)的范围,提升第1方式的方法的通用性的观点出发,优选空的交联微囊的交联率低。空的交联微囊的交联率越高,能够维持空的交联微囊的单分散性的最大施加外力越大,另一方面,空的交联微囊的交联率越低,能够维持空的交联微囊的单分散性的最大施加外力越小。因此,向空的交联微囊施加的外力越大,交联率的下限越高,另一方面,向空的交联微囊施加的外力越小,交联率的下限越低。例如,与通过搅拌的混合相比超声波照射的混合方法,由于施加在空的交联微囊的外力大,交联率的下限变高。从扩大在向空的交联微囊封入目标物质时能够使用的混合方法的范围,提升第1方式的方法的通用性的观点出发,优选空的交联微囊的交联率高。在将重量平均分子量10000~40000的目标物质封入空的交联微囊的时,使用搅拌作为混合的方法的情况下,交联率的下限,优选为35%,更优选为37%,进一步优选为38%,在使用超声波照射作为混合方法的情况下,交联率的下限优选为50%,更优选为55%。在任意的情况下,交联率的上限优选为80%,更优选为75%。交联率的测定方法可以根据交联剂的种类适当选择。在1-(3-二甲氨基丙基)-3-乙基碳二亚胺(EDC)等氨基缩合型的交联剂的情况下,作为交联率计算方法的一个例子,可以列出,基于由红外吸收光谱求出的羧基(-COO-)的消耗量,测定交联率的方法。另外,作为其他方法,可以列出,基于酰胺键(-COONH-)的增加量,测定交联率的方法。但是,在第1以及第2带电嵌段为聚氨基酸或其衍生物的情况下,由于第1以及第2带电嵌段含有大量的酰胺键,因此有降低测试精度的风险。因此,在这种情况下,优选和其他的测试方法(例如,基于羧基消耗量的测试方法)组合使用。进一步地,作为其他方法,可以列出,基于通过氨基染色剂定量的氨基残存量,计算出交联率的方法。作为氨基染色剂,可以列出,例如,三硝基苯磺酸,荧光胺等。在由交联膜包围的空间(空隙部)中,通常作为内水相含有在制备空的交联微囊的单分散体时使用的水性介质。[空的交联微囊的单分散体的制备方法]作为空的交联微囊的单分散体的制备方法,可以列出,例如,将空的非交联微囊的单分散体与能与第1和/或第2聚合物反应的交联剂反应的方法。构成空的非交联微囊的单分散体的各微囊为含有第1以及第2聚合物的,包括第1和/或第2聚合物都未交联成的非交联膜,和由非交联膜包围成的内水相的,在内水相中不包含目标物质的微囊。构成空的非交联微囊的单分散体的各微囊的结构如上述[B2-1:空微囊的结构]中记载。空的非交联微囊的单分散体的平均粒径,通常为30nm以上,优选为50nm以上,更优选为70nm以上,通常为10000nm以下,优选为1000nm以下,更优选为400nm以下。空的非交联微囊的单分散体的制备方法,如上述[B2-4:空微囊的制备方法]中记载。交联剂并不限定于能够与第1和/或第2聚合物反应。交联剂可以单独使用1种,也可以组合使用2种以上,根据使用交联剂的种类,形成选自由第1聚合物间的交联键、第2聚合物间的交联键、以及第1聚合物和第2聚合物间的交联键组成的组中的1种或2种以上的交联键。应该指出,在交联键中,参与交联反应的交联剂可以残存,也可以不残存。例如,1-(3-二甲氨基丙基)-3-乙基碳二亚胺(EDC),如后所述,在交联键中不残存。能够和第1和/或第2聚合物反应的交联剂中,包括,具有2个以上能够和第1聚合物具有的官能团反应的官能团,能够在第1聚合物间交联的交联剂(以下称为“第1交联剂”),具有2个以上能够和第2聚合物具有的官能团反应的官能团,能够在第2聚合物间交联的交联剂(以下称为“第2交联剂”),以及,具有1个以上能够与第1聚合物具有的官能团反应的官能团和1个以上能够与第2聚合物具有的官能团反应的官能团,能够在第1聚合物和第2聚合物间交联的交联剂(以下称为“第3交联剂”),从提升形成的物质包封交联微囊的单分散性的观点出发,优选单独使用或与其他交联剂组合使用第3交联剂。第1~第3交联剂,优选能够与第1和/或第2聚合物具有的官能团中的带电嵌段的带电基团反应。即,第1交联剂,优选能够在第1带电嵌段具有的带电基团间交联,第2交联剂,优选能够在第2带电嵌段具有的带电基团间交联,第3交联剂,优选能够在第1带电嵌段具有的带电基团和第2带电嵌段具有的带电基团间交联。能够和第1~第3交联剂反应的阳离子基团,优选为氨基。能够和第1~第3交联剂反应的阴离子基团,例如,羧基、磷酸基等,优选为羧基。能够和第1~第3交联剂反应的官能团,可以是在中性条件下不带电荷,在特定的条件下具有电荷的官能团,作为这样的官能团,可以列出,例如苯酚衍生物,吡啶衍生物,咪唑衍生物,硫醇衍生物等。这些官能团,也能够被设计为在中性条件下具有电荷的分子。作为交联氨基的交联剂,可以列出,例如,戊二醛、二甲基辛二硝酸二盐酸脂(DMS)、3,3’-二硫基二亚胺代丙酸二甲酯(DTBP)、双琥珀酰亚胺戊二酸脂(DSG)、辛二酸二琥珀酰亚胺(DSS)等琥珀酰亚胺衍生物、1,5-二氟-2,4-二硝基苯(DFDNB)等氟苯衍生物、3-[三(羟甲基)膦酰]丙酸(THPP)、双马来酰亚胺衍生物等。作为氨基和羧基缩合交联(因此交联剂在交联键中不残存)的交联剂,可以列出,例如,1-乙基-3-(3-二甲氨基丙基)碳化二亚胺(EDC)、N,N'-二环己烷碳二亚胺(DCC)、二异丙基碳二亚胺、N,N'-羰基咪唑(CDI)等的碳二亚胺系缩合剂、、4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉氯化物n水合物(DMT-MM)等。作为交联氨基以及磷酸的交联剂,可以列出EDC和咪唑复合系交联剂等。作为交联磷酸基团的交联剂,可以列出例如钙离子等金属离子等。理想的交联剂为EDC。EDC在交联键中不残存。因此,在将微囊给药到生物体内的情况下(例如,作为药物传递用微囊在生物体内给药的情况),即使微囊在生物体内被分解,也不显示出交联剂游离的细胞毒性。交联剂的使用量,以达到所期望的交联率的方式,可以根据交联剂的种类、交联对象成分的量、目标物质的种类、向空的交联微囊封入目标物质时使用的混合方法的种类等进行适当调节。在将重均分子量10000~40000的目标物质封入空的交联微囊的时,若假设使用交联氨基和羧基的交联剂(EDC等)作为交联剂的情况,交联剂的使用量优选为以下的范围。即,在封入时通过搅拌混合使用的情况下,上式(iii)规定的CL比,优选为0.3以上,更优选为0.5以上,而且,优选为1.2以下,更优选为1.0以下,调节交联剂的使用量。在封入时使用超声波混合使用的情况下,上式(iii)规定的CL比,优选为0.8以上,更优选为1.0以上,而且,优选为4.0以下,更优选为3.0以下调节交联剂的使用量。[混合液]混合液同时含有目标物质和水性介质。目标物质为应包封于微囊内的物质,可以根据微囊的用途等适当选择。目标物质如上述[B3:被包封物质]中的记载,水性介质如上述[B1-3:混合相关的其他条件]中的记载。混合液中含有的目标物质的浓度,在第1和/或第2聚合物不交联方面将与空的交联微囊的单分散体不同的空的非交联微囊的单分散体共同在混合液中混合的情况下,为阻碍内水相中包封有目标物质的物质包封非交联微囊的单分散体形成的浓度。构成空的非交联微囊的单分散体的各微囊为含有第1以及第2聚合物的,包含第1和/或第2聚合物都未交联成的非交联膜,和由非交联膜包围成的内水相的,内水相中不包含目标物质的微囊。构成物质包封非交联微囊的单分散体的各微囊为含有第1以及第2聚合物的,包含第1和/或第2聚合物都未交联成的非交联膜,和由非交联膜包围成的内水相的,内水相中包含目标物质的微囊。空的非交联微囊的单分散体在第1和/或第2聚合物不交联方面与空的交联微囊不同。以是否形成物质包封微囊的单分散体仅和交联的有无有关的方式,空的非交联微囊的单分散体,在交联以外的方面,优选和空的交联微囊的单分散体实质上是相同的。另外,以是否形成物质包封微囊的单分散体仅和交联的有无有关的方式,将空的非交联微囊的单分散体在混合液中混合时的条件(混合液的组成、混合方法等)优选和将空的交联微囊的单分散体在混合液中混合时的条件实质上是相同的。将空的非交联微囊的单分散体,在目标物质存在下,混合在水性介质的情况下,有在混合液中含有的目标物质的浓度超过一定浓度时,就会阻碍物质包封微囊的单分散体的形成(即,形成多分散体)情况。该一定浓度,根据目标物质的种类而不同。例如,目标物质的分子量为10000~40000的情况下,通常为5mg/mL、优选为15mg/mL、更优选为40mg/mL以上的浓度。在目标物质是难溶于水的物质的情况下,越增加混合液中含有的难溶于水的物质的浓度,第1方式的方法的效果越显著。即,在将空的非交联微囊的单分散体混合在混合液的情况下,当混合液中含有的难溶于水的物质的浓度超过一定浓度时,有阻碍物质包封非交联微囊的单分散体形成的情况(即,形成多分散聚合物)。对此,在将空的交联微囊的单分散体混合在混合液的情况下,即使超过该一定浓度,也不会阻碍物质包封交联微囊的单分散体的形成。因此,混合液中含有的难溶于水的物质的浓度,在将空的非交联微囊混合在同一混合液中的情况下,优选阻碍物质包封非交联微囊的单分散体形成的浓度。这样的浓度,通常比难溶于水物质的溶解度高(此时,难溶于水物质在混合液中分散,悬浊)。应该指出,“难溶于水”意味着在水性介质中难溶或不溶,例如,在水性介质为水的情况下,意味着相对于25℃的水中的溶解度通常为1.0mg/mL以下,优选为0.1mg/mL以下,更优选为0.02mg/mL以下。混合液中混合的空交联微囊的浓度,可以在微囊不凝聚的范围内进行适当调节。当空的交联微囊的浓度过低时,由于在混合后有不形成物质包封微囊情况,因此空的交联微囊的浓度优选在微囊不凝聚的范围内的高浓度。空的交联微囊的浓度,通常为0.1mg/mL以上,优选为1mg/mL以上,更优选为10mg/mL以上,通常为100mg/mL以下,优选为70mg/mL,更优选为50mg/mL。应该指出,由于可以认为获得的物质包封微囊的粒径和空的交联微囊的浓度有关,因此空的交联微囊的浓度应该根据要制备的物质包封微囊的粒径决定。混合液的pH没有特别的限制,可以根据空的交联微囊的结构、目标物质的种类、混合液中空的交联微囊以及目标物质的浓度等进行适当调整,但优选为pH5以上,更优选为pH6.5以上,另外优选为pH9以下,更优选为pH7.5以下。pH的调整可以通过使用作为溶剂的缓冲液容易地进行。混合液的pH调整后使用的操作,有利于保持空的交联微囊的结构,有效地将目标物质包封于空的交联微囊内。混合液的盐浓度(离子强度),能够在不破坏空微囊的结构、不阻碍向空的交联微囊中的目标物质的包封的范围内进行适当调整。但是,第1方式的方法,越是增加混合液的盐浓度(离子强度),其效果越显著。即,在将空的非交联微囊的单分散体在混合液中混合的情况下,当混合液的盐浓度超过一定值时,有阻碍物质包封非交联微囊的单分散体形成的情况(即,形成多分散体)。对此,在将空的交联微囊的单分散体在混合液中混合的情况下,即使超过该一定值,也不会阻碍物质包封交联微囊的单分散体的形成。因此,混合液的盐浓度,在将空的非交联微囊在同一混合液中混合的情况下,优选阻碍物质包封非交联微囊的单分散体形成的浓度。混合液的盐浓度,用氯化钠的浓度换算,优选为20mM以上,更优选为75mM以上,进一步优选为150mM以上。应该指出,混合液的盐浓度的上限,通常为1.0M,优选为0.5M。混合液的粘度,能够在不破坏空的交联微囊的结构、不阻碍向空的交联微囊中的目标物质的包封的范围内进行适当调整。但是,第1方式的方法,越是增加混合的粘度,其效果越显著。即,在将空的非交联微囊的单分散体在混合液中混合的情况下,当混合液的粘度超过一定值时,就会有阻碍物质包封非交联微囊的单分散体形成的情况(即,形成多分散体)。对此,在将空的交联微囊的单分散体在混合液中混合的情况下,即使超过该一定值,也不会阻碍物质包封交联微囊的单分散体的形成。因此,混合液的粘度,在将空的非交联微囊在同一混合液中混合的情况下,优选阻碍物质包封非交联微囊的单分散体形成的粘度。混合液的粘度,优选为0.05Pa.s以上,更优选为0.1Pa.s以上,进一步优选为0.5Pa.s以上。应该指出,混合液的粘度的上限通常为1.0Pa.s,优选为为0.75Pa.s。混合液的温度如果是在不破坏空微囊的结构、不阻碍向空的交联微囊中的目标物质的包封的范围内没有限制,优选为4℃以上,更优选为20℃以上,另外,优选为80℃以下,更优选为50℃以下。[混合方法]混合方法如上述[B1-2:空微囊以及被包封物质的混合]中的记载。在目标物质为难溶于水的物质的情况下,优选使用超声波处理作为混合方法。[其他的工序]第1方式的方法除了将空的交联微囊的单分散体在同时含有目标物质和水性介质的混合液中混合,形成在内水相中包封有目标物质的物质包封交联微囊的单分散体的工序以外,还可以包括其他的工序。例如,作为后工序可以包括针对将空的交联微囊在混合液中混合获得的物质包封交联微囊的交联工序。交联工序中使用的交联剂为能够和第1和/或第2聚合物反应的交联剂,其详细内容和上述记载相同。[物质包封交联微囊的单分散体]根据第1方式的方法,制备含有第1以及第2聚合物的,包含第1和/或第2聚合物交联成的交联膜,和交联膜包围成的内水相的,在内水相中包含有目标物质的物质包封交联微囊的单分散体。物质包封交联微囊的单分散体中,内水相中含有的目标物质的浓度,在第1和/或第2聚合物未交联方面以及目标物质未包封的方面,将与物质包封交联微囊的单分散体不同的空的非交联微囊的单分散体在同时含有与物质包封交联微囊的内水相同浓度是目标物质和水性介质的混合液中混合的情况下,为阻碍在内水相中包封有目标物质的物质包封非交联微囊的单分散体形成的浓度。换而言之,根据第1方式的方法制备的物质包封交联微囊具有比将空的非交联微囊的单分散体在同时含有与物质包封微囊的内水相同浓度的目标物质和水性介质的混合液中混合而获得的物质包封非交联微囊高的单分散性。物质包封微囊的内水相根据第1方式的方法中使用的混合液而变化。例如,在混合液的盐浓度以氯化钠浓度换算为20mM以上的情况下,内水相的盐浓度也相同。另外,在混合液的粘度为0.05Pa·s以上的情况下,内水相的粘度也相同。另外,混合液在以5mg/mL以上浓度含有重均分子量10000~40000的目标物质的情况下,内水相也相同。另外,混合液在以比其溶解度高的浓度含有难溶于水的物质的情况下,内水相也相同。物质包封交联微囊的交联率根据在第1方式的方法中使用的空的交联微囊的交联率,以及,作为后处理而实施的交联工序的有无而变化。例如,空的交联微囊中,第1和/或第2聚合物是通过选自由阳离子基团间形成的交联键、阴离子基团间形成的交联键、以及阳离子基团和阴离子基团间形成的交联键组成的组中的1种或2种以上的交联键交联而成,在形成的交联键的比例为交联膜中含有的阳离子基团和/或阴离子基团的总摩尔数的35%以上的情况下,将空的交联微囊在混合液中混合获得的物质包封交联微囊的交联率变成和空的交联微囊相同,在实施作为后处理的交联工序的情况下,相比于空的交联微囊,交联率有所增加。<C-2:第2方式/多元物质包封交联微囊及其制备方法>[概要]第2方式的方法为制备多元物质包封交联微囊的方法,包括将包封第1目标物质的第1物质包封交联微囊在同时含有比第1目标物质分子量小的第2目标物质和水性介质的混合液中混合,形成包封第1以及第2目标物质的第2物质包封交联微囊的工序。将包封第1目标物质的第1物质包封交联微囊在第2目标物质存在下,在水性介质中混合,在维持第1目标物质的封入的同时,将第2目标物质封入第1物质包封交联微囊中。因此,根据第2方式的方法,可以将第1以及第2目标物质控制封入量地封入,据此,可以制备控制了第1以及第2目标物质的封入量的物质包封微囊。[第1物质包封交联微囊]第1物质包封交联微囊为含有第1以及第2聚合物的,包含第1和/或第2聚合物交联成的交联膜和由交联膜包围成的内水相的,在内水相中包封第1目标物质的微囊。第1物质包封交联微囊的结构除了目标物质为第1目标物质以外,和第1方式的方法的物质包封交联微囊的结构相同。第1物质包封交联微囊优选为其单分散体。通过使用其单分散体作为第1物质包封交联微囊,可以将第2物质封入交联微囊作为其单分散体制备。第1物质包封交联微囊的单分散体的平均粒径通常为30nm以上,优选为50nm以上,更优选为70nm以上,通常为10000nm以下,优选为1000nm以下,更优选为400nm以下。[第1物质包封交联微囊的制备方法]作为第1物质包封交联微囊的制备方法,可以列出,例如,将含有第1以及第2聚合物的,包含第1和/或第2聚合物交联成的交联膜和由交联膜包围成的内水相的,在内水相中均不含有第1以及第2目标物质的空的交联微囊,在同时含有第1目标物质和水性介质的混合液中混合,根据需要,混合后,和能够与第1和/或第2聚合物反应的交联剂反应的方法(以下称为方法1。),将含有第1以及第2聚合物的,包含第1和/或第2聚合物都未交联的非交联膜和由非交联膜包围成的内水相的,在内水相中包封第1目标物质的物质包封非交联微囊,和能够与第1和/或第2聚合物反应的交联剂反应的方法(以下称为方法2。)等。方法1除了使用第1目标物质作为目标物质以外,可以和第1方式的方法相同地实施。方法2中使用的物质包封非交联微囊,可以是例如,通过同时混合法、后负载法等制备的物质。同时混合法为将第1聚合物、第2聚合物以及目标物质在水性介质中混合的方法,后负载法为将空微囊在同时含有第1物质和水性介质的混合液中混合的方法。方法2中使用的交联剂和在第1方式的方法中使用的交联剂相同。通过方法1以及方法2的任一方法,能将第1物质包封交联微囊作为其单分散体制备。在使用方法1作为第1物质包封交联微囊的制备方法的情况下,空的交联微囊优选为其单分散体,第1物质包封交联微囊优选为其单分散体。在使用其单分散体作为空的交联微囊的情况下,如果混合液中含有的第1目标物质的浓度在第1和/或第2聚合物未交联的方面将与空的交联微囊的单分散体不同的空的非交联微囊的单分散体在相同混合液中混合的情况下下,为阻碍在内水相中包封有第1目标物质的物质包封非交联微囊的单分散体形成的浓度,则能够取得和第1方式的方法相同的效果。[混合液]混合液同时含有比第1目标物质分子量小的第2目标物质和水性介质。第2目标物质,除了比第1目标物质分子量小,没有特别的限制,可以根据微囊的用途等进行适当选择。作为第1以及第2目标物质的具体例,可以列出和在第1方式的方法中使用的目标物质相同的具体例。作为第2目标物质理想的例子,可以列出拥挤试剂。拥挤试剂导致分子拥挤的环境(分子拥挤的环境)。拥挤试剂为不和第1目标物质相互作用且虽然导致分子拥挤环境但具有充分的溶解度的水溶性分子,作为其具体例,可以列出,聚乙二醇、糖的聚合物(葡聚糖、Ficoll(注册商标))、白蛋白蛋白质等亲水性高分子、甘油、乙二醇、二甘醇等亲水性小分子等。水性介质和在第1方式的方法中使用的水性介质相同。混合液的组成、pH、盐浓度(离子强度)、粘度等和在第1方式的方法中使用的混合液相同。在使用其单分散体作为空的交联微囊的情况下,如果混合液中含有的第2目标物质的浓度在第1和/或第2聚合物未交联方面将与第1物质包封交联微囊的单分散体不同的第1物质包封非交联微囊的单分散体在同混合液中混合的情况下下,为阻碍在内水相中包封有第1以及第2目标物质的第2物质包封非交联微囊的单分散体形成的浓度,则能取得和第1方式的方法相同的效果。[混合方法]混合方法和第1方式的方法相同。[物质包封交联微囊]根据第2方式的方法,制备含有第1以及第2聚合物的、包括由第1和/或第2聚合物交联成的交联膜、和由交联膜包围成的内水相的,在内水相中包封有第1以及第2目标物质的物质包封交联微囊。作为包封有第1以及第2目标物质的物质包封交联微囊的应用,可以列出,通过使用第2目标物质达到分子拥挤环境等,使第1目标物质稳定化。即,在物质包封交联微囊的一种方式中,第1目标物质相比于第2目标物质不存在包含在内水相中的情况是稳定化的。这样的第1目标物质的稳定化可以通过控制第2目标物质的封入量实现。此时,优选使用拥挤试剂作为第2目标物质。通过使用拥挤试剂作为第2目标物质,例如,可以提升第1目标物质的pH稳定性。[D]吸附材料包封微囊及其制备方法本发明的别的方式,公开了包封吸附材料的微囊(适当地称为“吸附材料包封微囊”)及其制备方法。本发明人等锐意研究的结果,发现,通过在具有相反电荷的2种聚合物的自组装的静电相互作用型微囊中,包封具有物质吸附能力的粒子(吸附材料粒子),能够获得包封相对于微囊的大小比较大的粒子的新型物质包封微囊(吸附材料包封微囊)。而且,发现,通过将2种聚合物的一方和吸附材料粒子混合,然后和另一方的聚合物混合,可以极其有效地获得这样的吸附材料包封微囊。本方式是基于这样见解的发现。本发明的吸附材料包封微囊包括由含有具有不带电亲水性嵌段以及第1带电嵌段的嵌段聚合的第1聚合物,以及具有带与所述第1带电嵌段相反电荷的第2带电嵌段的第2聚合物的膜形成的微囊,和在该微囊内包封的吸附材料粒子,所述第1以及第2聚合物的至少一方被吸附在所述吸附材料粒子上。其中,关于第1以及第2聚合物如上述[B]栏中说明。本说明书中的吸附材料粒子是指具有物质吸附作用的粒子。其中吸附是指在两相(这里为固体粒子和其周围的液相)的界面处,物质的浓度相比于周围较高的现象。吸附粒子具有的吸附作用的种类没有限制,可以是物理吸附(通过物理的相互作用吸附),也可以是化学吸附(伴随着化学键的吸附),也可以是它们组合。在物理吸附的情况下,作为其基础的物理的相互作用,可以列出,离子间相互作用、偶极相互作用、范德华力(VanderWaals)等、或者它们2种以上的组合,任何作用都可以。在化学吸附的情况下,作为其基础的化学键,可以列出,氢键、共价键、配位键、离子键等、或者它们2种以上的组合,任何作用都可以。但是,作为吸附材料粒子优选为表面具有电荷的对水性介质表现分散性的粒子。可以认为通过吸附材料粒子具有这些性质,作为微囊构成要素的第1和/或第2聚合物容易通过静电相互作用吸附在吸附材料粒子上,促进在吸附材料粒子表面附近的微囊的形成。具体而言,粒子表面Zeta电位的绝对值通常为10mV以上,优选为20mV以上,更优选为30mV以上。应该指出,粒子表面的Zeta电位可以使用例如光散射电泳法等的电泳法进行测定。此外,从提升吸附能力的观点出发,吸附粒子优选为比表面积大的粒子。作为比表面积大的粒子可以列出各种多孔性粒子。另外,在多孔性粒子的情况下,粒子的孔容为0.1cm3/g以上,优选0.5cm3/g以上。应该指出,粒子表面的比表面积或多孔性粒子的孔容等的孔隙特征可以使用例如BET法等进行测定。吸附材料粒子的种类没有限制,可以列出,例如,二氧化硅、磷酸钙、碳酸钙、氧化铝、沸石、氧化铁等各种金属氧化物;金、铂、钯等金属单质;聚苯乙烯树脂、苯乙烯-二乙烯基苯树脂、离子交换树脂、聚丙烯酸粒子、聚乳酸颗粒、乳酸-羟基乙酸共聚物微粒等合成或天然树脂等的材料形成的粒子。其中,优选二氧化硅、磷酸钙、金、聚丙烯酸微粒等。此外,吸附材料粒子的性状也没有限制,可以是固体粒子,也可以在水性介质中形成胶体或凝胶。进一步地,也可以是最初不是吸附材料或粒子状物质,包封到微囊内后能够成为吸附材料粒子的物质。有关这样的形态的例子,稍后叙述。吸附材料粒子的尺寸也没有限制,本发明中,由于能够包封在现有的静电相互作用型微囊中难以包封的大粒径(例如超过30nm)的粒子,因此优选使用这样的大粒径的吸附材料粒子。其中,优选为具有通常40nm以上,尤其50nm以上,进一步地60nm以上的直径的吸附材料粒子。平均粒径的上限也没有限制,通常10μm以下的程度。应该指出,吸附材料粒子的平均粒径取决于吸附材料的种类,但通常可以根据动态光散射(dynamiclightscattering:DLS)、透射电子显微镜(transmissionelectronmicroscope:TEM)、扫描电子显微镜(scanningelectronmicroscope:SEM)、激光衍射法、库尔特计数法等方法测定。根据本发明的吸附材料包封微囊,能够包封相对于微囊比较大的吸附材料粒子。例如,根据后述的实施例1,相对于作为包封对象的吸附材料粒子(介孔二氧化硅纳米粒子:MSN)的平均粒径约为80nm,获得的吸附材料包封微囊的平均粒径约为100nm,虽然吸附材料粒子的粒径相对微囊粒径极其大,但能获得以高包封率地将其包封的吸附材料包封微囊。其原因并不明确,但可以考虑是否是由于第1和/或第2聚合物在吸附在吸附材料粒子上的状态下进行自组装,在吸附材料粒子的周围有效地形成微囊。本发明的吸附材料包封微囊,能够通过将第1以及第2聚合物,连同吸附材料粒子以任意的顺序在通常是水性介质中混合制备。但是,从提升向吸附材料粒子的微囊的包封率的观点出发,优选通过至少包括(a)在水性介质中,预先将第1以及第2聚合物的任一方和吸附材料粒子混合,使其被吸附材料粒子吸附的工序,以及,(b)在含有水性介质的该混合中进一步加入其他的聚合物进行混合,在吸附材料粒子的周围形成由含有第1以及第2聚合物的膜形成的微囊的工序的方法(本发明的吸附材料粒子包封微囊的制备方法)进行制备。通过这样的制备方法,能够大幅提高吸附材料粒子的包封率,能高效率地制备所期望的吸附材料粒子包封微囊。其中,工序(a)中预先吸附到吸附材料粒子的聚合物(第1或第2聚合物),可以为任何聚合物。但是,从吸附材料包封微囊的制备效率等观点出发,优选将具有与吸附材料粒子的表面电荷相反电荷的聚合物预先与吸附材料粒子接触。例如,在吸附材料粒子的表面电荷为正的情况下,优选为具有负电荷的聚合物(即,具有阴离子性嵌段的聚合物),在吸附材料粒子的表面电荷为负的情况下,优选为具有正电荷的聚合物(即,具有阳离子性嵌段的聚合物)。应该指出,在吸附材料粒子的表面上正负电荷混在一起的情况下,综合考虑吸附剂粒子全体的表面电荷判断正负即可。然而同样的,无论哪一个聚合预先与吸附材料粒子接触,都能够获得被发明的吸附材料包封微囊。关于水性介质的种类和,第1以及第2聚合物以及吸附材料粒子的各浓度、混合方法、其他的制备的的各种条件,能够将上述[B]栏中说明的条件直接、或适当加以变更使用。根据以上的制备方法,在吸附材料粒子的周围形成由含有第1以及第2聚合物的膜形成的微囊,制得吸附材料包封微囊。其中,在使用带电粒子作为吸附材料粒子的情况下,吸附材料包封微囊,和未包封在微囊内的吸附材料粒子,可以通过例如表面Zeta电位等进行识别。即,相对于带电的吸附材料粒子的表面Zeta电位的绝对值大,在经由微囊包封吸附材料粒子的吸附材料包封微囊中,通过第1以及第2聚合物的静电相互作用,电荷略微消失,表面Zeta电位的绝对值变小。因此,确认表面Zeta电位的绝对值降低,进而考虑通过DLS测定的粒子物性和TEM观察等的结果,能够证实吸附材料包封微囊的形成。根据上述制备方法,吸附材料粒子的包封率增加的理由没有定论,但推测如下。即,本制备方法中,不是将吸附材料粒子包封在自组装后的微囊中,而是通过在作为包封对象的吸附材料粒子上吸附第1以及第2聚合物的一方后,加入另一方,在吸附材料粒子的表面附近形成微囊的膜。换而言之,可以认为,在微囊的形成时,由于将吸附材料粒子作为模板使用,通过有选择地增高在吸附材料粒子附近的微观环境中的带电聚合物(第1以及第2聚合物中的一方)的浓度(认为是吸附的一种),在加入具有相反电荷的聚合物(第1以及第2聚合物中的另一方)时,在吸附材料粒子的表面附近有选择地发生自组装。其结果,可以将通常仅能按对应溶液中数密度的概率封入的物质,以极其高的包封效率包封。另外,在包封尺寸比较大的吸附材料粒子的情况下,通过同样的理由,可以认为由于各种聚合物在吸附材料粒子的表面以被吸附的状态进行自组装,在吸附材料周围有效地形成微囊,包封成为可能。应该指出,本发明的吸附材料包封微囊的制备方法中,可以加入上述以外的其他工序实施。作为例子,在吸附材料包封微囊形成后优选将微囊中的第1和/或第2聚合物交联。通过交联,能够提高微囊的稳定性。通过将聚合物交联,例如,在DDS中使用吸附材料包封微囊的情况下,即使在生理条件下,也能够维持微囊的稳定性。应该指出,有关将第1和/或第2聚合物交联的方法,将单独描述。此外,在吸附材料包封微囊的形成前、形成时、或形成后,可以实施将吸附剂表面处理的工序。通过进行表面处理,能够在吸附剂表面赋予各种官能团,能够改变吸附剂表面的各种特性。表面处理的种类没有特别的限制,例如,在使用二氧化硅作为吸附材料的情况下,可以列出通过各种硅烷偶联剂的处理等。硅烷偶联剂的种类也没有特别的限制,作为例子,在表面赋予阳离子性基团的情况下,可以通过例如3-氨基丙基三甲氧基硅烷、3-氨基丙基三乙氧基硅烷等氨基化硅烷偶联剂进行处理。另外,例如在表面赋予阴离子性基团的情况下,可以通过例如3-巯基丙基三甲氧基硅烷、3-巯基丙基三乙氧基硅烷等巯基化硅烷偶联剂处理后,将巯基氧化,转换为磺酰基。其他的,可以列出通过可以期待芳香环的疏水性和π电子相互作用等的p-苯乙烯基三甲氧基硅烷、乙烯基三甲氧基硅烷、乙烯基三乙氧基硅烷、3-丙烯酰基三甲氧基硅烷、3-甲基丙烯酰基丙基三甲氧基硅烷等的表面处理。当然,表面处理并不限定在这些处理中,可以根据所期望的特性和用途、使用吸附材料的种类、吸附材料中吸附的物质(后述)的种类等,适当选择任意的表面处理。此外,本发明中,微囊中包封的吸附材料粒子也能够吸附别的物质。特别地,即使有关在现有的静电相互作用型微囊中难以稳定包封的,小分子量(例如5000Da以下等)的化合物,根据本发明,以在吸附材料中吸附负载的状态,能够稳定地包封在微囊中。据此,本发明的吸附材料包封微囊能够在负载缓释各种药剂等的小分子化合物的用途中适宜地使用。特别地,通过使用如上所述在表面具有电荷、针对水性介质显示出分散性的粒子作为吸附材料粒子,能够在生理条件下稳定存在的同时,能够在生理条件下缓释吸附材料中负载的各种药剂等小分子化合物,能够实现优异的DDS。另外,根据负载药剂等小分子化合物的种类,通过对吸附剂实施如上所述的表面处理,能够提高其吸附负载特性。小分子化合物向吸附材料粒子吸附负载,在吸附材料粒子包封在微囊前后都可以,由于通过被吸附的小分子化合物的物理化学的性质,吸附材料粒子的物理化学性质也受到影响,可以认为,将吸附材料粒子包封在微囊后的方法为理想的。应该指出,作为本形态的变形例,通过调制导入了能够形成吸附材料的物质(吸附材料前体)的物质包封微囊,其后将吸附材料前体转变为吸附材料,也能够形成吸附材料包封微囊。作为吸附材料前体的例子,可以列出,能够形成凝胶等的基质或沉淀、胶束等的带电聚合物等。此时,调制导入了带电聚合物物质包封微囊,其后,在该微囊内导入相反电荷的药剂等。据此,静电相互作用适当地取消,带电聚合物疏水化,在微囊内部凝胶等的基质或沉淀、胶束等的吸附材料形成的同时,形成在该基质、沉淀、胶束等吸附材料中捕获药剂的药物负载吸附材料包封微囊。这样的药物负载吸附包封微囊作为缓释该药剂的缓释剂也是有用的。[E]低水溶性物质包封微囊及其制备方法本发明的另外的方式公开了能够制备包封低水溶性物质的微囊的物质包封微囊的制备方法,和通过该方法制备的新型低水溶性物质包封微囊。本发明人等锐意研究的结果,发现,通过准备能够把相比于目标物质水溶性高的前体转变为目标物质的酶包封在微囊中的酶包封微囊,使前体向酶包封微囊内渗透,通过微囊内的酶,将其转变为目标物质,使目标物质析出并包封在微囊内,能够使效率的包封成为可能。本形态是基于这样见解的发现。本方式的物质包封微囊的制备方法为在由包含作为具有不带电亲水性嵌段以及第1带电嵌段的嵌段聚合物的第1聚合物,以及,具有带与第1带电嵌段相反电荷的第2带电嵌段的第2聚合物的膜形成的微囊中,包封目标物质的物质包封微囊的制备方法,包括以下工序。(a)将能够将相比于所述目标物质水溶性高的前体转变为所述目标物质的酶,包封在含有所述第1以及第2聚合物的膜形成的微囊中的准备酶包封微囊的工序。(b)在相比于所述前体,对于所述目标物质表现低溶解度的条件下,向所述酶包封微囊内,渗透所述前体,通过所述酶将所述前体转变为所述目标物质,使所述目标物质析出包封在所述酶包封微囊内,作为低水溶性物质包封微囊的工序。本形态中,微囊内包封的目标物质为能够通过酶由水溶性高的前体转变得到的物质。除了存在相比于目标物质水溶性高的前体,而且存在能够使前体向目标物质转变的酶以外,可以利用任意种类的物质作为目标物质。换而言之,本形态中,微囊内包封的目标物质为通过酶作用降低在水中溶解度的物质。此时,受到酶作用之前的在水中溶解度相对高的状态的物质成为前体,受到酶作用之后的在水中溶解度相对低的状态的物质成为目标物质。因此,作为前体和目标物质的组合,不仅化学式和化学组成不同的物质的组合,而且即便化学式和化学组成相同,但通过立体结构等变化在水中溶解度变化的物质也能成为对象。本方式中,优选目标物质的溶解度对于前体的溶解度的比率小。具体而言,虽不限于此,但在比较相对于25℃水中溶解度的情况下,目标物质的溶解度对于前体的溶解度的比率,优选通常为90%以下,尤其是80%以下,进一步地为70%以下。此外,也根据前体以及目标物质各自的水溶性或用途,但优选前体和目标物质的水溶性的差别大。具体而言,在比较相对于25℃水中溶解度的情况下,和目标物质相比,前体一方具有优选通常10mg/mL以上,尤其20mg/mL以上,进一步地30mg/mL以上高的溶解度的情况。此外,优选目标物质为低水溶性。具体而言,相对于25℃的水中溶解度,通常为1.0mg/mL以下,优选为0.3mg/mL以下,更优选为0.1mg/mL以下。作为上述定义的目标物质以及前体,和媒介其转变的酶的组合,各种组合都是已知的,本发明可以使用其中任意组合。作为一个例子,如果考虑制备的微囊在DDS等使用,可以列出例如通过各种酶转变成活性药物的各种酶前体药物体系等。本形态中,首先,准备在由含有第1以及第2聚合物的膜形成的微囊中包封有酶的酶包封微囊。这样的酶包封微囊使用上述[B]栏中说明的各种物质包封微囊的制备方法(后负载法、同时混合法等),和上述[C]栏中说明的物质包封微囊的制备方法等任意方法,通过在微囊内包封酶进行调制。然后,在相比于前体目标物质表现低溶解性的条件下,向酶包封微囊内渗透前体。向酶包封微囊内渗透前体的方法是任意的,通常通过将酶包封微囊和前体在溶剂中混合实施。这种情况下,可以是分别调制在各种溶液中含有酶包封微囊和前体的溶液后,将它们混合,也可以是向在溶剂中含有酶包封微囊以及前体中一方的溶液直接添加另一方。进一步地,也可以在液体等的连续相使酶包封微囊和前体接触(例如后述的在体内(invivo)使用符合这样的形态)。其中,所谓的相比于前体目标物质表现低溶解性的条件为虽然前体能够以大致溶解在溶剂中的状态存在但目标物质的部分或全部从溶剂中析出的条件。通过考虑前体以及目标物质的各自水溶性选择适当的溶剂,可以实现。例如,在目标物质为上述定义的低水溶性物质,前体为上述定义的水溶性物质的情况下,通过一般的水性介质,可以的实现这样的条件。另外,通过在溶剂中溶解各种有机或无机的电解质调节溶解度,或调节温度等各种环境因素,控制溶剂的溶解度,也可以实现该条件。通过在上述条件下向酶包封微囊内渗透前体,由酶将前体向目标物质转变。其中,优选使得通过酶从前体到目标物质是转变反应确切地进行,选择与目标物质/前体/酶的组合对应的适当的条件。这样通过由酶将前体向目标物质转变,目标物质从溶剂中析出并包封在酶包封微囊内,形成物质包封微囊。关于其他的制备方法中的各种条件,能够直接或加以适当变更地使用上述[B]栏中说明的条件。应该指出,本形态的物质包封微囊的制备方法中,也可以增加上述以外的其他工序后实施。作为例子,可以列出,向酶包封微囊内渗透前体前和/或后,增加交联第1和/或第2聚合物的工序。特别地,从防止酶从酶包封微囊中脱落,微囊稳定化的观点出发,在向酶包封微囊内渗透水溶性前体前,优选将酶包封微囊内的第1和/或第2聚合物交联。通过聚合物的交联,例如,在DDS等使用本方式的情况下,即使在生理条件下也能够维持微囊的稳定。应该指出,有关将第1和/或第2聚合物交联的方法,能直接或加以适当变更地使用上述[C]栏中说明的条件。根据本方式的物质包封微囊的制备方法,可以认为由于使用预先将酶封入微囊内的酶包封微囊,可以将目标物质的生成场所限制为仅在微囊内部,通过这样,能够有效地将目标物质包封在微囊中。其结果,可以将通常只能根据溶液中的数密度对应的概率封入的物质,以极其高的包封效率包封。进一步地,通过变化向酶包封微囊中添加的前体的量,也可以控制目标物质向微囊的包封量。并且,可以认为由于微囊具有半透膜是性质,能够从微囊缓释微囊中包封的目标物质。此外,根据本形态的物质包封微囊的制备方法,即使是有关现有的难以或是不可能向微囊包封的低水溶性物质,也能够将其形成包封的微囊。即,根据本方式,在由含有前述第1以及第2聚合物的膜形成的微囊中,低水溶性物质(和酶一同)被包封,能够制得低水溶性物质包封微囊。这样的低水溶性物质包封微囊也是本发明的主旨。这样的微囊中,低水溶性物质优选以超过内水相中低水溶性物质溶解度的浓度进行包封。另外,这样的微囊中优选前述第1和/或第2聚合物交联。此外,可以将通过本方式的制备方法的物质包封微囊的形成在体外(invitro)人为地实现。通过适当地选择目标物质/前体/酶的组合等,也能以在体外实现的方式进行设计。有关这样的形态,将在后面叙述。[F]其他以上,参照各种方式对本发明进行了说明,但是本发明并不限定在以上的形态中,能够增加任意的变形后实施。例如,上述<C-1>中说明的物质包封交联微囊的单分散体、上述<C-2>中说明的多元物质包封交联微囊、上述<D>中说明的吸附材料包封微囊、上述<E>中说明的低水溶性物质包封微囊,通过负载药剂,能用作为医药组合物或药物传递系统使用。含有这样的本发明的各种物质包封微囊的医药组合物或药物传递系统也是本发明的对象。适用这样的医药组合物或药物传递系统的对象(任意生物,但优选为动物,更优选为哺乳动物,进一步优选为人)、疾病、药物等可以适当选择。另外,这样的医药组合物或药物传递系统的给药途径、用法、用量等各种条件可以根据对象、疾病、药物等进行适当选择。此外,可以将通过上述[E]中说明的制备方法的物质包封微囊的形成在体外人为地实现,但通过适当地选择目标物质/前体/酶的组合等,也可以以在体内实现的方式进行设计。即,向体内导入在血液或体液等生理溶液中表现溶解性(水溶性)的前体,同时通过其他路径向体内导入酶包封微囊,在体内所期望的部位,使酶包封微囊和前体接触,前体向酶包封微囊渗透。这样通过将前体转变为生理溶液中表现低溶解性(低水溶性)的目标物质,可以在体内形成含有该目标物质的物质包封微囊。其中,作为目标物质以及前体的组合,如果采用如上所述的药物和其酶前体药物的组合,在体内所期望的的器官或组织等部位,前体药物向药物转变并包封在微囊内,从形成的药物包封微囊缓释药物,能够实现优异的DDS。而且,将具有将药物的前体(前体药物)转变为药剂作用的酶作为上述[C]的物质包封交联微囊的被包封物质,调制包封的物质包封微囊(酶包封交联微囊)、或将该酶吸附、负载于上述[D]的吸附材料包封微囊的吸附材料上的物质包封微囊(酶负载吸附材料包封微囊),也可以使用这些微囊。在这种情况下,将前体药物导入体内的同时,通过其他途径将这些酶包封交联微囊或酶负载吸附材料包封微囊向体内导入,在体内所期望的部位,使酶包封交联微囊或酶负载吸附材料微囊和前体药物接触,使前体药物向微囊渗透。这样通过酶对向前体药物作用转变为药物,在体内可以由前体药物生成药物。应该指出,这样的方式,不仅是本发明的各种物质包封微囊,使用任意的物质包封微囊都能够实现。即,调制使用包封负载酶的任意微囊(酶负载微囊)。在这种情况下,将前体药物导入体内的同时,通过其他途径将所述的酶负载微囊向体内导入,在体内所期望的部位,使酶负载微囊和前体药物接触,使前体药物向微囊渗透。这样通过酶对前体药物作用转变为药物,在体内可以由前体药物生成药物。在这些方式中,药物和其前体药物的溶解度的关系没有限制,能够使用任意的药物/前体药物/酶的组合。作为这样的药物/前体药物/酶的组合的例子,可以列出下表所示的组合,但这些只是示例,作为本发明对象的药物/前体药物/酶的组合并不限定在这些组合中。[表1]即,根据本发明,为用于向对象传递药物的方法,进一步提供了含有以下工序的方法。(a)由含具作为有不带电亲水性嵌段以及第1带电嵌段的嵌段聚合物的第1聚合物,以及,具有带与前述第1带电嵌段相反电荷的第2带电嵌段的第2聚合物的膜形成的微囊中,包封能将所述药物的前体(前体药物)转变为所述药物的酶的准备酶包封微囊的工序。(b)在对象的预定部位,通过使所述的前体向所述酶包封微囊内渗透,借助所述酶使所述前体转变为所述药物的形成所述药物的工序。上述方法的工序(b)在对象的预定部位中,通过酶包封微囊和前体共存而实现。对象的预定部位是任意的,可以根据传递药物的对象、药物的种类、作为治疗对象的疾病等决定,但通常为对象预定的器官、组织或细胞。在对象的预定部位使酶包封微囊存在的方法是任意的,但通常通过现有公知的各种给药方法,将酶包封微囊向对象给药,能够到达对象的预定部位即可。给药方法可以是口服也可以是非口服,在非口服给药的情况下,可以以静脉内、肌内、皮下、经皮、经鼻、经肺等任何途径给药。上述方法的工序(b)中,通过酶包封微囊内包封的酶,将前体转变为药物即可。据此,转变的药物从微囊缓释,传递到对象预定的部位。但是,从将药物长期地稳定地传递的观点出发,优选药效析出后包封在酶包封微囊中,形成药物包封微囊。其他的用量和用法等的各种条件可以根据药物传递的对象,药物的种类,作为治疗对象的疾病等适当地决定。此外,使用以上方法(含有酶包封微囊等)的药物传递系统和医药组合物也是本发明的对象。实施例下面,参照实施例,更加具体地说明本发明。应该指出,以下实施例,仅是作为示例的目的,对本发明没有任何限制。应该指出,在以下的记载中,除非另有说明,所谓“溶液”或“分散液”是指以10mM磷酸缓冲液(pH7.4)作为溶剂或分散剂的溶液或“分散液”。此外,以下的记载中,除非另有说明,“旋涡混合器”使用Eppendorf社制的MixMate。此外,以下记载中,“平均粒径”、“多分散指数(polydispersityindex:PDI)”以及“Zeta电位”,除非另有说明,通过Malvern社制的ZetasizerNano-ZS进行测定。此外,以下的记载中,“透射电子显微镜图像”,除非另有说明,是使用日本电子制JEM-1400取得的。[实施例群I]物质包封微囊及其制备方法实施例I-1向空的交联微囊封入FITC-Dex40k(1)空微囊的调制使用由不带电亲水性嵌段的聚乙二醇(重均分子量约2000)(以下有表示为“PEG”的情况)和阴离子性嵌段的聚天冬氨酸(聚合度约75)形成的阴离子性嵌段聚合物PEG-P(Asp),作为第1聚合物。部分聚天冬氨酸的N末端通过Cy3进行荧光标记。使用由聚(含有两个氨基结构的天冬酰胺衍生物)(聚合度约82)(以下有表示为“P(Asp-AP)”的情况)形成的阳离子性均聚物Homo-P(Asp-AP),作为第2聚合物。将第1以及第2聚合物分别以聚合物浓度为1.0mg/mL的方式溶解在10mM磷酸缓冲液(pH7.4)(水性介质)中。将获得的第1聚合物的溶液以及第2聚合物的溶液以相等电荷比(即C/A比=1.0)的方式加入微量离心管中进行混合,使用旋涡混合器,2000rpm下搅拌2分钟,获得含有通过第1以及第2聚合物的自组装而形成的微囊(空微囊)的溶液。对获得的空微囊含有液,通过动态光散射法(dynamiclightscattering)进行测定,求出粒径分布、平均粒径以及多分散指数(PDI)。其结果,证实了平均粒径为101.4nm、PDI为0.070的单分散性微囊粒子的形成。(2)空微囊的交联将0.5mL上述获得的空微囊含有液,加到含有相对于PEG-P(Asp)中羧基的0.5倍等量EDC(同仁化学研究所制)的溶液(pH7.4的10mM磷酸缓冲液(0mMNaCl)中),室温下静置12小时,获得空微囊交联后的空的交联微囊含有液。(3)向空的交联微囊封入FITC-Dex40k使用异硫氰酸荧光素-葡聚糖(Sigma-Aldrich制,以下有表示为“FITC-Dex40k”的情况)(重均分子量40000),作为被包封物质。向0.5mL的上述获得的空的交联微囊含有液中加入FITC-Dex40k溶液(混合后,FITC-Dex40k浓度为5~50mg/mL、带电聚合物(第1以及第2聚合物)的浓度为1.0mg/mL),使用旋涡混合器,2000rpm下搅拌2分钟,向空的交联微囊中封入FITC-Dex40k,获得含有FITC-Dex40k包封交联微囊的溶液。获得的溶液为透明的。向1.0mL以充分的外力施加条件(2000rpm下搅拌2分钟)进行混合得到的溶液中,加入含有相对于PEG-P(Asp)中含有的羧基的10倍等量EDC的溶液(pH7.4的10mM磷酸缓冲液(0mMNaCl)中),室温下静置12小时,使FITC-Dex40k包封交联微囊充分交联。将未包封在微囊中在溶液中以游离状态残存的FITC-Dex40k通过离心超滤(使用VIVASPIN20,SartoriusStediumBiotech社制,截止分子量为30万)(离心超滤条件:300rpm,4℃)除去进行精制后,通过动态光散射法(dynamiclightscattering)进行测试,求出粒径分布、平均粒径以及多分散指数(PDI)。其结果,FITC-Dex40k包封交联微囊的粒径分布、平均粒径以及多分散指数(PDI),和空微囊的这些性质基本没有变化,证实了在空微囊中,维持其单分散性的同时,封入了FITC-Dex40k。例如,和50mg/mL的FITC-Dex40k溶液搅拌混合得到的FITC-Dex40k包封交联微囊的平均粒径为97.2nm,PDI为0.087(空微囊的平均粒径为101.4nm,PDI为0.070),透射电子显微镜(TEM)图像如图5A所示。接下来,室温下,通过卡尔蔡司社制的激光共聚焦荧光显微镜(LSM510)进行荧光相关光谱(FCS)测试。荧光葡萄糖的激发为氩激光器(480nm)、物镜使用40倍的浸水镜头,数据分析通过Confocor3软件求出自相关函数G(τ)的值。根据G(τ)的值,估算每个FITC-Dex40k包封交联微囊中FITC-Dex40k的个数。应该指出,自相关函数G(τ)为评价时间τ后的荧光强度波动的函数,随时间衰减,经过充分的时间后汇聚。到汇聚为止经过的时间越长(衰减晚),波动越缓,即,表示测试对照的荧光物体的运动迟钝。另外,由于观察视野内含有的分子数越增加,波动减小,从相关函数的值可以估算分子数。每个FITC-Dex40k包封交联微囊的FITC-Dex40k个数如表2所示。如表2所示,每个FITC-Dex40k包封交联微囊的FITC-Dex40k个数,随着与空微囊混合的FITC-Dex40k溶液浓度的上升而增加。[表2]从以上的结果,发现,将空微囊的单分散体交联后,通过在FITC-Dex40k溶液中,施加充分外力条件下混合,维持其单分散性的同时,向空微囊中封入FITC-Dex40k,制备FITC-Dex40k包封微囊的单分散体。此外,从实施例I-1和比较例I-1,I-2(后述)的比较,发现,即使在水性介质中存在的FITC-Dex40k的浓度超过5mg/mL,能形成FITC-Dex40k包封微囊的单分散体的,仅有实施例I-1.比较例I-1通过同时混合法封入FITC-Dex40k和实施例I-1相同,将第1以及第2聚合物分别以聚合物浓度为2.0mg/mL的方式溶解在10mM磷酸缓冲液(pH7.4)(水性介质)中。此时,向第1聚合物溶液中加入预定的FITC-Dex40k,加入FITC-Dex40k量要保证,在和第2聚合物的溶液混合后FITC-Dex40k的最终浓度为5~50mg/mL,带电聚合物(第1以及第2聚合物)最终浓度为1.0mg/mL。将获得的第1聚合物/FITC-Dex40k混合液和第2聚合物的溶液,以第1聚合物以及第2聚合物相等电荷比(即C/A比=1.0)的方式加入微量离心管中混合,使用旋涡混合器,2000rpm下搅拌2分钟,获得含有第1以及第2聚合物的自组装聚合体的溶液。向0.5mL所得溶液中添加含有相对于PEG-P(Asp)中羧基的10倍等量EDC的溶液(pH7.4的10mM磷酸缓冲液(0mMNaCl)中),室温下静置12小时。将溶液中以游离状态残存的FITC-Dex40k通过和实施例I-1相同的离心超滤去除后,通过DLS法和TEM,评价获得的自组装聚合体。如图5B所示,发现在FITC-Dex40k最终浓度为50mg/mL的情况下获得的自组装聚合体为单层微囊,但粒径有显著增大的倾向。从DLS的结果来看,也可以确认尺寸增大、单峰消失、以及PDI显著增大(平均粒径:约630nm,PDI:约0.62)。这可以认为是由于微囊形成过程受到溶液粘度的影响,给与了宽的粒径分布。另一方面,在FITC-Dex40k最终浓度为5mg/mL的情况下获得的自组装聚合体,形成了粒径为100nm程度的单分散微囊粒子,可以通过FCS估算FITC-Dex40k封入量(表2)。[比较例I-2]通过后负载法封入FITC-Dex40k和实施例I-1相同,将第1以及第2聚合物分别以聚合物浓度为1.0mg/mL的方式溶解在10mM磷酸缓冲液(pH7.4)(水性介质)中。将获得的第1聚合物的溶液以及第2聚合物的溶液按照第1聚合物和第2聚合物相等电荷比(即C/A比=1.0)的方式加入到微量离心管中混合,使用旋涡混合器,2000rpm下搅拌2分钟,获得含有第1以及第2聚合物的自组装聚合体的溶液。根据DLS的结果,平均粒径为101.8nm、PDI为0.081。向0.5mL所得溶液中加入预定的FITC-Dex40k溶液,使混合后FITC-Dex40k的最终浓度为5~50mg/mL,带电聚合物(第1以及第2聚合物)的浓度为1.0mg/mL,再次使用旋涡混合器,2000rpm下搅拌2分钟。接着,加入含有相对于PEG-P(Asp)中羧基的10倍等量EDC的溶液(pH7.4的10mM磷酸缓冲液(0mMNaCl)中),室温下静置12小时。将溶液中以游离状态残存的葡聚糖通过和实施例I-1相同的离心超滤去除后,通过DLS法和TEM,评价获得的自组装聚合体。如图5C所示,发现在FITC-Dex40k最终浓度为50mg/mL的情况下获得的自组装聚合体为单层微囊,但粒径有显著增大的倾向。从DLS的结果来看,也可以确认尺寸增大、单峰消失、以及PDI显著增大(平均粒径:约560nm,PDI:约0.57)。另一方面,在FITC-Dex40k最终浓度为5mg/mL的情况下获得的自组装聚合体形成了粒径为100nm程度的单分散微囊粒子,可以通过FCS估算封入量(表2)。[参考例I-1]从FITC-Dex40k包封微囊中释放FITC-Dex40k和实施例I-1相同,将第1以及第2聚合物分别以聚合物浓度为1.0mg/mL的方式溶解在10mM磷酸缓冲液(pH7.4)(水性介质)中。将获得的第1聚合物的溶液以及第2聚合物的溶液按照第1聚合物和第2聚合物相等电荷比(即C/A比=1.0)的方式加入到微量离心管中混合,使用旋涡混合器,2000rpm下搅拌2分钟,获得含有第1以及第2聚合物的自组装聚合体的溶液。向0.5mL所得溶液中加入预定的FITC-Dex40k溶液,使混合后FITC-Dex40k的最终浓度为4mg/mL,带电聚合物(第1以及第2聚合物)的浓度为1.0mg/mL,再次使用旋涡混合器,2000rpm下搅拌2分钟。接着,加入含有10倍等量EDC的溶液(pH7.4的10mM磷酸缓冲液(0mMNaCl)中),室温下静置12小时。将未被微囊包封的在溶液中以游离状态残存的葡聚糖通过和实施例I-1相同的离心超滤去除。精制后的FITC-Dex40k包封微囊在37℃,150mMNaCl的条件下静置。对微囊内封入的FITC-Dex40k的释放行为使用GPC(凝胶渗透色谱,日本分光制)进行确认。如图6所示,在刚精制后和3日后,在色谱图上没有发现显著的差别,没有证实FITC-Dex40k的释放。从该结果推测,在不给于混合等外力的情况下,对于微囊不会发生FITC-Dex40k的出入。[参考例I-2]FITC-Dex浓度和粘度的关系比较例1中,暗示了微囊形成过程(特别是搅拌混合情况)受到溶液粘度的影响,因此,研究了FITC-Dex(重均分子量4000,10000,40000)的浓度和粘度的关系。粘度的测试,使用旋转流变仪(AntonPaar社制PhysicaMCR302)粘度(Pa.s)的测试结果如表3所示。[表3][实施例I-2]向细胞色素c包封交联微囊封入FITC-Dex4k(1)细胞色素c包封交联微囊的调制和比较例I-2同样进行,施行向空的微囊内细胞色素c(Sigma-Aldrich社制,分子量12327Da)的封入。其具体的方法,如下所述。将第1以及第2聚合物分别以聚合物浓度为2.0mg/mL的方式溶解在10mM磷酸缓冲液(pH7.4)(水性介质)中。将获得的第1聚合物的溶液以及第2聚合物的溶液按照第1聚合物和第2聚合物相等电荷比(即C/A比=1.0)的方式加入到微量离心管中混合,使用旋涡混合器,2000rpm下搅拌2分钟,获得含有通过第1以及第2聚合物的自组装而形成的微囊(空微囊)的溶液。从DLS的结果来看,平均粒径为102nm、PDI为0.045。向0.96mL所得的空微囊含有液中加入0.5mL的1mg/mL细胞色素c溶液,再次使用旋涡混合器,2000rpm下搅拌2分钟后,获得细胞色素c包封微囊含有液。向0.5mL所得的细胞色素c包封微囊含有液中加入含有相对于PEG-P(Asp)中羧基的0.5倍等量EDC的溶液(pH7.4的10mM磷酸缓冲液(0mMNaCl)中),室温下静置12小时,使细胞色素c包封微囊交联,获得细胞色素c包封交联微囊含有液。将未包封在微囊中的在溶液中以游离状态残存的细胞色素c通过和实施例I-1相同的离心超滤(截止分子量为30万)除去,精制微囊。此时,平均粒径为99.5nm,PDI为0.032.根据GPC测试结果,证实了微囊内封入了细胞色素c(最大吸收波长409nm)(图7)。应该指出,图7中,(a)表示空微囊的GPC测试结果,(b)表示精制后细胞色素c包封交联微囊的GPC测试结果。(2)向细胞色素c包封交联微囊封入FITC-Dex4k向0.5mL所得的细胞色素c包封交联微囊含有液中加入FITC-Dex4k溶液(重均分子量:4000,浓度:50mg/mL,容量:0.5mL),使用旋涡混合器,2000rpm下搅拌2分钟,细胞色素c包封交联微囊中封入FITC-Dex4k,调制细胞色素c以及FITC-Dex4k包封交联微囊含有液。向细胞色素c以及FITC-Dex4k包封交联微囊含有液中加入含有相对于具有PEG-P(Asp)的羧基的10倍等量EDC的溶液(pH7.4的10mM磷酸缓冲液(0mMNaCl)中),室温下静置12小时,使细胞色素c以及FITC-Dex4k包封交联微囊充分交联。将未包封在微囊中的在溶液中以游离状态残存的FITC-Dex4k通过和实施例I-1相同的离心超滤除去。根据DLS测试的结果,平均粒径为101nm,PDI为0.078。根据微囊的吸收光谱,微囊中封入了细胞色素c以及FITC-Dex4k,即,证实了在向细胞色素c包封交联微囊中封入FITC-Dex4k时,没有释放细胞色素c地封入FITC-Dex4k(图8)。应该指出,图8中,(a)表示细胞色素c包封交联微囊的吸收光谱,(b)表示细胞色素c以及FITC-Dex4k包封微囊的吸收光谱。将FITC-Dex4k溶液的浓度在1~100mg/mL内变化,和上述相同,调制细胞色素c以及FITC-Dex4k包封交联微囊,和实施例I-1相同地进行FCS测试,估算每个微囊的FITC-Dex4k个数。其结果取表4所示。正如表4所示,每个微囊的FITC-Dex4k个数也随着FITC-Dex4k浓度的上升而增加。[表4]然后,基于参考文献(K.Sasahara,etal.J.Mol.Biol.(2003)326,1227-1237),通过圆二色谱,评价在酸性环境中细胞色素c的立体结构的稳定性。根据参考文献,细胞色素c的立体结构自pH2.0将丢失,特征的α-螺旋线结构也丢失。另一方面,在高浓度的葡聚糖存在的情况下,可以防止其结构的消失。根据圆二色谱(图9),在Dex没有共存在微囊内的情况下,当pH降低到2.0时,222nm附近的吸收消失,相对于此,在Dex共存于微囊内的情况下,没有观察到显著的变化。其结果,可以认为,通过在微囊内创造出Dex高浓度共存的分子拥挤环境,可以取得效果。应该指出,图9中,“(A)CytC@PICsome(pH7.4)”表示pH7.4中细胞色素c包封交联微囊的圆二色谱,“(B)CytC@PICsome(pH2.0)”表示pH2.0中细胞色素c包封交联微囊的圆二色谱,“(C)(CytC+FITC-Dex)@PICsome(pH7.4)”表示pH7.4中细胞色素c以及FITC-Dex4k包封交联微囊的圆二色谱,“(D)(CytC+FITC-Dex)@PICsome(pH7.4)”表示pH7.4中细胞色素c以及FITC-Dex4k包封交联微囊的圆二色谱。[实施例I-3]向空的交联微囊封入AlPcS2a向与实施例I-1相同调制的空微囊中加入相对于含有PEG-P(Asp)的羧基的1.0倍等量的EDC,制备空的交联微囊。此时,平均粒径为101nm,PDI为0.084。调制不同浓度(0、1、2、10、20、50mg/mL)的磺化酞菁(AlPcS2a)(FrontierScientific社制)分散液,并以微囊浓度为1mg/mL的方式向空的交联微囊中混合,施行2分钟的超声波照射。应该指出,微囊浓度通过荧光标识Cy3定量。AlPcS2a为光动力疗法使用的药剂(光增感剂),具有以下结构。[化学式10]超声波处理后,为了除去溶液中以游离状态残存的AlPcS2a,通过PD-10柱(GE医疗社制)以及离心超滤(VIVASPIN20,SartoriusStediumBiotech社制,截止分子量使用30万)进行精制。精制后的微囊,根据DLS测试,不论AlPcS2a浓度,都维持其平均粒径为110nm程度,PDI为0.05~0.08程度的粒径、单分散性。另外,对精制后的微囊进行基于AlPcS2a的吸收色的比色实验。如图10(A)所示,发现随着接触的AlPcS2a浓度越高,由AlPcS2a而来的蓝色变强。[比较例I-3]和比较例I-2相同,将第1以及第2聚合物分别以聚合物浓度为2.0mg/mL的方式溶解在10mM磷酸缓冲液(pH7.4)(水性介质)中。将获得的第1聚合物的溶液以及第2聚合物的溶液按照第1聚合物和第2聚合物相等电荷比(即C/A比=1.0)的方式加入到微量离心管中混合,使用旋涡混合器,2000rpm下搅拌2分钟,获得含有第1以及第2聚合物的自组装聚合体的溶液。向0.5mL所得溶液中加入预定的AlPcS2a溶液,使混合后AlPcS2a的最终浓度为2.0mg/mL,带电聚合物的浓度为1.0mg/mL,再次使用旋涡混合器,2000rpm下搅拌2分钟。接着,加入含有相对于含PEG-P(Asp)的羧基的10倍等量EDC的溶液(pH7.4的10mM磷酸缓冲液(0mMNaCl)中),室温下静置12小时。将未包封于微囊中的在溶液中以游离状态残存的AlPcS2a通过和实施例I-1相同的离心超滤去除。根据DLS测试,平均粒径为104nm,PDI为0.062。通过将封入的AlPcS2a的量在图10(A)中表示的比色实验,和实施例I-3的结果相比,发现,实施例I-3的一方在接触相同浓度的AlPcS2a的情况下封入了更多的量。另外,在混合后的AlPcS2a浓度比2.0mg/mL大的情况下难以维持粒径和PDI。此外,将实施例I-3以及比较例I-3的微囊溶液(AlPcS2a浓度均为20mg/mL)稀释至带电聚合物浓度为0.5mg/mL,使用分光光度计测定吸收光谱。其结果如图10(B)所示。发现还是实施例I-3的一方,与比较例I-3相比,在接触相同浓度的AlPcS2a的情况下封入了更多的量。[参考例I-3]NaCl浓度对空微囊的影响使用由不带电亲水性嵌段的聚乙二醇(重均分子量约2300)(以下有表示为“PEG”的情况)和阴离子性嵌段的聚天冬氨酸(聚合度约79)组成的阴离子性嵌段聚合物PEG-P(Asp)作为第1聚合物。使用由聚(含有两个氨基结构的天冬酰胺衍生物)(聚合度约71)(以下有表示为“P(Asp-AP)”的情况)组成的阳离子性均聚物Homo-P(Asp-AP)作为第2聚合物。将第1以及第2聚合物分别以聚合物浓度为1.0mg/mL的方式溶解在10mM磷酸缓冲液(pH7.4;水性介质)中。将获得的35.5μL第1聚合物的溶液和50μL第2聚合物的溶液以相等电荷比(即C/A比=1.0)的方式加入到微量离心管中混合,使用旋涡混合器,2000rpm下搅拌2分钟,获得含有通过第1以及第2聚合物的自组装而形成的微囊(空微囊)的溶液。其中,加入含有预定量NaCl的10mM磷酸缓冲液,使最终浓度为5、10、20、150mM、静置一晚。对于获得的空微囊含有液通过动态光散射法(dynamiclightscattering)进行测试,求出粒径分布,平均粒径、多分散指数(PDI)以及Zeta电位。NaCl添加前,平均粒径为126nm,PDI为0.049的空微囊在分别添加NaCl后显示出如表5的值。特别地,在20mMNaCl存在下,PDI显示出约为0.2的极大的值,TEM图像也证实了含有大量的直径超过200nm的粒径的成分的多分散性微囊的形成。另外,在添加150mMNaCl的情况下,空微囊溶液直接浑浊,暗示了尺寸以及多分散性的增大。[表5]接着,使用由不带电亲水性嵌段的聚乙二醇(重均分子量约2300)(以下有表示为“PEG”的情况)和阴离子性嵌段的聚天冬氨酸(聚合度约79)组成的阴离子性嵌段聚合物PEG-P(Asp)作为第1聚合物。使用由聚(含有两个氨基结构的天冬酰胺衍生物)(聚合度约71)(以下有表示为“P(Asp-AP)”的情况)组成的阳离子均聚物Homo-P(Asp-AP)作为第2聚合物。将第1以及第2聚合物分别以聚合物浓度为1.0mg/mL的方式溶解在10mM磷酸缓冲液(pH7.4;制备3种含有无NaCl、10、20mMNaCl的情况;水性介质)中。将获得的第1聚合物的溶液和第2聚合物的溶液和前项同样地以相等电荷比(即C/A比=1.0)的方式加入到微量离心管中混合,使用旋涡混合器,2000rpm下搅拌2分钟,获得通过第1以及第2聚合物的自组装而形成的粒子。通过动态光散射评价粒径及其分布,获得了和前项相同倾向的结果(表6),在20mMNaCl存在下,PDI为0.2,暗示了多分散性的提高。[表6]NaCl浓度(mM)粒径(nm)PDI01320.058101510.065201900.2[参考例I-4]交联率的决定使用由不带电亲水性嵌段的聚乙二醇(重均分子量约2000)(以下有表示为“PEG”的情况)和阴离子性嵌段的聚天冬氨酸(聚合度约75)组成的阴离子性嵌段聚合物PEG-P(Asp)作为第1聚合物。将阴离子性聚合物的末端用Cy3标识(将阴离子性聚合物数量的10%标识)。使用由聚(含有两个氨基结构的天冬酰胺衍生物)(聚合度约82)(以下有表示为“P(Asp-AP)”的情况)组成的阳离子性均聚物Homo-P(Asp-AP)作为第2聚合物。分别将其以1mg/mL的方式溶解到pD=7.4的D2O中。将两溶液按照电荷比相等的方式混合,进行Vortex混合(2000rpm,2分钟),调制空微囊含有液。添加在系统中含有的相对于Cy3-PEG-P(Asp)的羧基的10、5、1、0.5、0.1等量分的EDC溶液(D2O,pD=7.4),调制空的交联微囊含有液。使用截止分子量30万的离心超滤膜(Vivaspin,GE医疗制)进行精制。超滤条件为2000rpm,25℃×10次,重新添加D2O精制。使用100μL10mg/mL的空的交联微囊含有液(溶剂D2O),实施全反射红外分光法(日本分光制,FTIR-6300)。将通过全反射红外分光法获得的1550-1600cm-1的峰使用分析软件(IGORPro)进行分离,通过定量COO-对应的面积的减少,算出交联率(表7)。[表7][参考例I-5]向空的非交联微囊封入FITC-Dex40k向与实施例I-1相同调制的空微囊中,与实施例I-1相同地封入异硫氰酸荧光素-葡聚糖(重均分子量40000)(Sigma-Aldrich制,以下有表示为“FITC-Dex40k”的情况),对于获得的FITC-Dex40k包封非交联微囊,通过动态光散射法测试粒径分布、平均粒径、多分散指数(PDI)。其结果,FITC-Dex40k的浓度为10mg/mL时的平均粒径为107.4nm、PDI为0.067,FITC-Dex40k的浓度为15mg/mL时的平均粒径为109.2nm、PDI为0.070(空微囊的平均粒径为101.4nm,PDI为0.070),证实了在维持其单分散性的同时,向空微囊中封入了FITC-Dex40k。[参考例I-6]针对空的非交联微囊的超声波处理将与实施例I-1相同地调制的空微囊进行超声波处理(2分钟),对于超声波处理后的微囊,通过动态光散射法,测试粒径分布、平均粒径、多分散指数(PDI)。其结果,在冰浴中实施超声波处理的情况下,平均粒径为112.8nm,PDI为0.098,微囊溶液中未发现异常变化,不在冰浴中实施超声波处理的情况下,平均粒径为3606nm,PDI为0.301,微囊溶液变得白色浑浊。根据其结果,可以认为,针对微囊的超声波处理最好以微囊不被过度加热的方式实施(例如,非连续地实施超声波处理,连续实施超声波处理的情况下将微囊冷却)。[实施例I-4]胞嘧啶脱氨酶封入交联微囊的制备胞嘧啶脱氨酶封入交联微囊的调制及评价封入胞嘧啶脱氨酶(cytosinedeaminase:CD)(胞嘧啶脱氨酶硫酸铵悬浮液(Cytosinedeaminaseammoniumsulfatesuspension),CALZYME制)的交联微囊,按照以下的顺序制备。应该指出,CD为将5-氟胞嘧啶(以下适当略为“5-FC”)转变为5-氟尿嘧啶(以下适当略为“5-FU”)的酶。向与实施例I-1相同调制的空微囊中加入相对于PEG-P(Asp)中含有的羧基的1.0等量的EDC后交联,制备空的交联微囊。将500μL约3mg/mL的空的交联微囊的(PEG-P(Asp)换算)分散液和500μ胞嘧啶脱氨酶(CD)1mg的分散液混合,通过使用旋涡混合器(2000rpm),搅拌2分钟,将CD负载在交联微囊内。然后,加入相对于PEG-P(Asp)中含有的羧基的9.0等量的EDC,在4℃下经过一晚,进一步交联,通过4℃,2000rpm的离心下超滤(截止分子量30万,PBS中)进行精制,获得1mLCD封入交联微囊的分散液。对于空交联微囊以及CD封入交联微囊,通过动态光散射(DLS)法进行测定,求出平均粒径以及多分散指数(PDI)。其结果表示在下表8中。另外,CD封入交联微囊的透射电子显微镜照片如图12所示。[表8]平均粒径(nm)PDI空交联微囊98.60.045CD封入交联微囊96.80.092通过胞嘧啶脱氨酶封入交联微囊的由5-FC向5-FU的转变对通过CD封入交联微囊的由5-FC向5-FU的转变效率按照以下顺序评价。将0.15mL以上述顺序调制的CD封入交联微囊的分散液和0.15mL0.01mg/mL的5-FC溶液混合,通过液相色谱法连续地测试混合液中的5-FU的浓度。结果如图13的图表所示。通过封入交联微囊的胞嘧啶脱氨酶5-FC向5-FU转变。其转变速度(酶活性)为0.03U/mL。通过胞嘧啶脱氨酶封入交联微囊的酶活性的稳定性对通过CD封入交联微囊的酶活性的稳定性按照以下的顺序评价。将以上述顺序调制的CD封入交联微囊在10MmPBS(pH7.4,150mMNaCl)中,4℃下保存。在调制起0日、5日、7日以及14日的时间点,在和上述相同条件下与5-FC溶液混合,按照上述方法测定40分钟后的混合液中的5-FU浓度。结果如图14的图表所示。调制后酶活性至少维持了7日。通过胞嘧啶脱氨酶封入交联微囊的肿瘤治疗效果准备每组5只、共8组(组1~8)的移植了小鼠大肠癌细胞株C26的BALB/c小鼠(6周龄),对于各小组,将以上述顺序调制的CD封入微囊(0.5U/mL)、5-FC、5-FU、或PBS各200μL在下表9表示的时间静脉内给药。5-FC以及5-FU以对应小鼠体重10mg/kg(体重)或80mg/kg(体重)的方式进行调整。给药后,各小鼠的生存期间中,最长持续28日,测定肿瘤体积的同时,测定各小鼠的体重。肿瘤体积,使用游标卡尺测定肿瘤的最大直径以及最小直径,通过以下的计算式算出。V=(a2×b)/2V:肿瘤体积,a:最大直径,b:最小直径[表9]肿瘤体积的测试结果如图15所示。CD封入交联微囊以及5-FC给药组(组1:10mg/kg;组2:80mg/kg),和非处置组(组8)比较,无论5-FU用量,都证实了显著性的抗肿瘤效果(图15(a))。将CD封入交联微囊以及5-FC给药组(组1:10mg/kg;组2:80mg/kg)和5-FU给药组(组3:10mg/kg;组4:80mg/kg)的每个用量进行比较,用量为10mg/kg时,对比于5-FU给药组(组3)仅显示出非常微弱的抗肿瘤效果,证实了CD封入交联微囊以及5-FC给药组(组1)的显著的优异的抗肿瘤效果。用量为80mg/kg时,虽然5-FU给药组(组4)观察到肿瘤缩小的抗肿瘤作用,但对比于其副作用所造成的初次给药后17天的全部死亡,CD封入交联微囊以及5-FC给药组(组2)发挥抗肿瘤效果的同时,没有表现出副作用,给药28日后,除了1只以外全部存活(图15(c))。在CD封入交联微囊单独给药组(组5)以及5-FC单独给药组(组6:10mg/kg;组7:80mg/kg)中,和非处置组相比较没有发现显著的抗肿瘤效果以及毒性(图15(d))。体重变化的测试结果如图16所示。CD封入交联微囊以及5-FC给药组(组1:10mg/kg;组2:80mg/kg)中,给药28天后,相对体重不低于0.8(图16(a)),但其他的组都低于0.8或死亡。(图16(b)~(d))。生存数的合计结果如图17所示。CD封入交联微囊以及5-FC给药组(组1:10mg/kg;组2:80mg/kg)中,相比于在给药28天后除组2(80mg/kg)的1只死亡以外全部生存(图17(a)),在5-FU10mg/kg给药组(组3)中3只直到其他组中全部给药28天后死亡(图17(b)~(d))。胞嘧啶脱氨酶封入交联微囊的血中滞留性评价准备3只移植了小鼠大肠癌细胞株C26的BALB/c小鼠(7周龄),将200μL的以Cy5标识的上述CD封入交联微囊(0.5U/mL)静脉内给药。在给药后24小时以及72小时的时间点,基于Cy5标识,测试血中CD量。血中CD量的测试结果如图18的图表所示。根据CD封入交联微囊,给药起24小时后以及72小时后的任意时间,血中都残存了高浓度的CD量。因此,了解到CD封入交联微囊在血中滞留性方面是优异的。[比较例I-4]通过后负载法向非交联微囊导入CD向与实施例I-1相同调制的交联前的空微囊分散液中加入CD分散液,通过使用涡轮搅拌器2000rpm下搅拌混合2分钟,调制带电聚合物(第1以及第2聚合物)浓度约0.9mg/mL、混合后的CD最终浓度为1mg/mL的微囊分散液。对该分散液通过动态光散射(DLS)法进行测定,求出平均粒径以及多分散指数(PDI),但不能证实均一的粒子的形成,为白色浑浊的状态。[实施例群II]吸附材料微囊及其制备方法[实施例II-1]MSN包封微囊1材料:使用由不带电亲水性嵌段的聚乙二醇(PEG;分子量约2000)和阴离子性嵌段的聚天冬氨酸(P(Asp);聚合度约75)形成的阴离子性嵌段聚合物(PEG-P(Asp);Zeta电位-30.6mV)作为第1聚合物。使用由阳离子性嵌段的聚(含有两个氨基结构的天冬酰胺衍生物)(P(Asp-AP);聚合度约82)形成的阳离子性均聚物(Homo-P(Asp-AP);Zeta电位+16.3mV)作为第2聚合物。根据Kimetal.,Angew.Chem.Int.Ed.2008,47,8438-8441中记载的方法使用制备的介孔二氧化硅纳米粒子作为被包封物质的吸附材料粒子。根据本文献,获得的介孔二氧化硅纳米粒子的总孔容为1.07cm3/g。此外,平均粒径约50nm,Zeta电位为-37mV。MSN包封微囊的调制以及评价将上述的第1以及第2聚合物分别溶解在10mM磷酸缓冲液(pH7.4)(水性介质)中,调制各聚合物浓度1.0mg/mL的溶液。另外,将上述的MSN通过超声波照射在10mM磷酸缓冲液(pH7.4)(水性介质)中分散,调制MSN浓度1.0mg/mL的分散液。在微量离心管中,向上述的第2聚合物的溶液中,以MSN的浓度成为总聚合物浓度(第1以及第2聚合物的总浓度)的20w/w%的方式添加上述MSN的分散液,用涡旋搅拌器2000rpm,搅拌2分钟,其后静置8分钟。向获得的微量离心管中的分散液中以和第2聚合物电荷比相等(即C/A比=1.0)的方式加入上述第1聚合物的溶液后混合,通过用涡旋搅拌器2000rpm,搅拌2分钟,获得含有第1以及第2聚合物自组装而形成的MSN包封微囊(吸附材料包封微囊)的溶液。对获得的MSN包封微囊含有液进行DLS测定,求出粒径分布、平均粒径以及PDI。证实了平均粒径为111nm的单分散粒子的形成。PDI为0.092。交联MSN包封微囊的调制及其评价将上述MSN包封微囊含有液加到含有相对于PEG-P(Asp)具有的羧基10等量EDC(同仁化学研究所制,WSC)的溶液中,静置一晚通过EDC反映进行交联。通过离心超滤(VIVASPIN20、sartoriusstediumbiotech社制,截止分子量30万;2000rpm,25℃)除去反应的副产物进行精制,获得含有交联的MSN包封微囊的溶液。对所得的交联MSN包封微囊含有液进行DLS测试,求出粒径分布、平均粒径以及PDI。粒径分布图如图19所示。证实了平均粒径为102nm的单分散粒子的形成。PDI为0.077,Zeta电位为-17mV。此外,对上述的交联MSN包封微囊含有液,通过透射电子显微镜(TEM,日本电子制JEM-1400,以下相同)也一并进行了形态观察,证实了DLS测试结果的准确性。获得的TEM照片如图20所示。MSN包封微囊(图中对比度高的粒子)的粒径为60~70nm,同时形成的空微囊(未包封MSN的粒子)的粒径约为100nm。没有观察到未封入的MSN。MSN包封率的测试此外,为了研究MSN包封微囊的MSN包封率,进行了以下实验。在MSN上使用吸附了异硫氰酸荧光素(Fluoresceinisothiocyanate:FITC)的FITC标识MSN,用和上述相同的方法,调制包含交联MSN包封微囊的溶液。对获得的交联MSN包封微囊含有液,通过毛细管电泳(BeckmancoulterInc.制P/ACEMDQ)进行测定。使用荧光检出器作为检出器,使用内径75μm的毛细管(polymicroTechnology製TSP075375)作为毛细管,使用200mM甘氨酸-氢氧化钠缓冲液(pH8.0)作为电泳液。MSN包封微囊和未包封在微囊内的MSN根据表面电荷的不同,通过毛细管电泳进行分离。获得的色谱图如图21所示。根据本结果,包封在微囊内的MSN为85%。血中滞留性的评价MSN包封微囊的血中滞留性,通过以下的顺序评价。首先,调制以下的样品(i)~(iv)。(i)Cy5标识MSN的样品:含有吸附荧光色素Cy5的MSN(Cy5相对MSN的比率为0.4%)的溶液。(ii)Cy5标识MSN包封微囊的样品:用上述(i)中使用的Cy5标识MSN代替MSN以外,通过和上述相同的顺序调制的MSN包封微囊含有液。(iii)MSN包封Cy5标识微囊的样品:使用Cy5标识的PEG-P(Asp)(Cy5相对PEG-P(Asp)的比率为10mol%)代替PEG-P(Asp)以外,通过和上述相同的顺序调制的,MSN包封微囊含有液。将上述(i)~(iv)的样品分别向BALB/c(雌,6周龄,每组各3只)尾静脉注射(PEG-P(Asp)换算浓度0.2mg/体重g)。1小时后进行采血,使用荧光分光器(日本分光制FP-7700)测试获得的血液中的荧光色素浓度。获得的结果(各组3只给药个体的平均值)如下表10所示。[表10][实施例II-2]MSN包封微囊2上述实施例II-1的顺序中,变更上述的第1以及第2聚合物的各自溶液与MSN分散液的混合顺序,首先将第1聚合物的溶液和MSN分散液混合,获得的混合液中混合第2聚合物的溶液以外,通过与实施例II-1相同的顺序,获得含有包封MSN微囊(MSN包封微囊:吸附材料包封微囊)的溶液。对获得的MSN包封微囊含有液,进行DLS测定,求出粒径分布、平均粒径以及PDI。证实了平均粒径为103nm的单分散粒子的形成。PDI为0.059。使用获得的MSN包封微囊含有液,通过与实施例II-1相同顺序进行交联,获得含有交联后的MSN包封微囊的溶液。对获得的交联MSN包封微囊含有液,进行DLS测定,求出粒径分布、平均粒径以及PDI。粒径分布图如图22所示。证实了平均粒径为101nm的粒子的形成。PDI为0.152。此外,对上述交联MSN包封微囊含有液,也通过TEM一并进行了形态观察,证实了DLS测试结果的准确性。获得的TEM照片如图23所示。MSN包封微囊(图中对比度高的粒子)的粒径为60~70nm,同时形成的空微囊(未包封MSN的粒子)的粒径约为100nm。此外,观察到少许未封入的MSN。此外,为了研究MSN包封率,以与实施例II-1相同的顺序,通过毛细管电泳进行测定。获得的色谱图如图24所示。根据本结果,包封在微囊中的MSN为72%。[参考例II-1]MSN包封微囊3上述实施例II-1的顺序中,变更上述的第1以及第2聚合物的各自溶液与MSN分散液的混合顺序,首先将第1聚合物的溶液和第2聚合物的溶液混合,向获得的混合液中混合MSN的分散液以外,通过与实施例II-1相同的顺序,获得含有包封MSN微囊(MSN包封微囊:吸附材料包封微囊)的溶液。对获得的MSN包封微囊含有液,进行DLS测定,求出粒径分布、平均粒径以及PDI。证实了平均粒径为104nm的单分散粒子的形成。PDI为0.049。使用获得的MSN包封微囊含有液,通过与实施例II-1相同顺序进行交联,获得含有交联后的MSN包封微囊的溶液。对获得的交联MSN包封微囊含有液,进行DLS测定,求出粒径分布、平均粒径以及PDI。粒径分布图如图25所示。证实了平均粒径为100nm的的粒子的形成。PDI为0.057。此外,对上述交联MSN包封微囊含有液,也通过TEM一并进行了形态观察,证实了DLS测试结果的准确性。获得的TEM照片如图26所示。MSN包封微囊(图中对比度高的粒子)的粒径为60~70nm,同时形成的空微囊(未包封MSN的粒子)的粒径约为100nm。此外,观察到部分未封入的MSN。此外,为了研究MSN包封率,以与实施例1相同的顺序,通过毛细管电泳进行测定。获得的色谱图如图27所示。根据本结果,包封在微囊中的MSN为21%。[实施例II-3A]胺化MSN包封微囊胺化MSN包封微囊的调制将50μL(3-氨基丙基)三甲氧基硅烷((3-Aminopropyl)trimethoxysilane:APTS),和4mL蒸馏水在常温下共同搅拌1小时溶解。接着,加入1mL10mg/mL的以与上述实施例II-1相同的顺序获得的MSN包封微囊(吸附材料包封微囊)的分散液,进一步搅拌24小时,对包封的MSN施行通过APST的表面处理。其后,离心超滤(截止分子量30万,20%乙醇用3次,其后10mMPBS用5次)精制,获得胺化MSN包封微囊。胺化MSN包封微囊的评价—平均粒径以及PDI对表面处理前的未处理MSN包封微囊以及胺化MSN包封微囊通过动态光散射(DLS)进行测定,求出平均粒径以及多分散指数(PDI)。其结果如下表11所示。另外,未处理MSN包封微囊以及胺化MSN包封微囊的透射电子显微镜照片分别如图28(a)以及(b)所示。[表11]胺化MSN包封微囊的评价—表面胺基此外,对未处理MSN包封微囊以及胺化MSN包封微囊,通过TNBS试验测定胺基的量。具体而言,将200μL由0.15M硼酸钠以及0.01M亚硫酸钠水溶液形成的缓冲液和50μL2,4,6-三硝基苯磺酸(2,4,6-trinitrobenzenesulfonicacid:TNBS)的0.1%水溶液,和50μL2mg/mL的未处理MSN包封微囊或胺化MSN包封微囊的分散液混合,37℃下放置一晚后,测试波长为420nm的紫外线(UV)吸光度。根据得到的吸光度,求出各MSN包封微囊中存在的胺基数。其结果,在胺化MSN包封微囊中,相比于未处理MSN包封微囊,每1mgMSN检测出了8.14×1019个多个的胺基。[实施例II-3B]巯基化以及磺酰化MSN包封微囊巯基化以及磺酰化MSN包封微囊的调制:将50μL(3-巯基丙基)三甲氧基硅烷((3-Mercaptopropyl)trimethoxysilane:MPTS),和4mL1%醋酸水溶液共同在常温下搅拌1小时溶解。接着,加入1mL10mg/mL的与上述实施例II-1相同顺序获得的MSN包封微囊(吸附材料包封微囊)的分散液,进一步搅拌24小时,对包封的MSN施行用过MPST的表面处理。其后,离心超滤(截止分子量30万,1%醋酸水溶液用3次,其后水用5次)精制,获得巯基化MSN包封微囊。接着,向该巯基化MSN包封微囊的分散液加入1mL30%过氧化氢水溶液以及10μL浓硫酸,通过12小时搅拌,将巯基转变为磺酰基。用5M氢氧化钠水溶液中和,用水稀释后,离心超滤(截止分子量30万,10mMPBS(pH7.4)用5次)精制,获得磺酰化MSN包封微囊。巯基化以及磺酰化MSN包封微囊的评价—平均粒径以及PDI对表面处理前的未处理MSN包封微囊、巯基化以及磺酰化MSN包封微囊,通过动态光散射(DLS)进行测定,求出平均粒径以及多分散指数(PDI)。其结果如下表12所示。另外,未处理MSN包封微囊以及磺酰化MSN包封微囊的透射电子显微镜照片分别如图29(a)以及(b)所示。[表12]平均粒径(nm)PDI未处理MSN包封微囊100.50.078巯基化MSN包封微囊106.60.122磺酰化MSN包封微囊109.10.113磺酰化MSN包封微囊的评价—MSN表面硫酸基此外,对未处理MSN包封微囊以及磺酰化MSN包封微囊,通过X射线分析显微镜(XGT-5200WR,(株)堀场制作所)证实了硫酸基的有无。获得的X射线分析谱图如图30所示。在磺酰化MSN包封微囊中确认了与硫酸基对应的S峰,与之相对,在未处理MSN包封微囊中,没有确认S峰。[实施例II-4A]孟加拉玫瑰红吸附胺化MSN包封微囊孟加拉玫瑰红吸附胺化MSN包封微囊的调制:向实施例II-3A中获得的胺化MSN包封微囊的分散液中加入孟加拉玫瑰红(rosebengal:以下适当略为“RB”)混合,在胺化MSN包封微囊中吸附孟加拉玫瑰红。通过超滤(截止分子量30万)除去未吸附的孟加拉玫瑰红。获得的RB吸附胺化MSN包封微囊的RB含有率为3.2w/w%。孟加拉玫瑰红的化学式如下所示。[化学式11]孟加拉玫瑰红吸附胺化MSN包封微囊的评价—释放特性对该RB吸附胺化MSN包封微囊,测定在10mMPBS(pH7.4,150mMNaCl)中37℃的孟加拉玫瑰红的释放特性。获得的结果如图31的图表所示。24小时内释放了约95%的孟加拉玫瑰红。[实施例II-4B]吉西他滨吸附磺酰化MSN包封微囊吉西他滨吸附磺酰化MSN包封微囊的调制:向实施例II-3B中获得的磺酰化MSN包封微囊的分散液中加入吉西他滨(gemcitabine:以下适当略为“GEM”)混合,在磺酰化MSN包封微囊中吸附吉西他滨。通过超滤(截止分子量30万)除去未吸附的吉西他滨。获得的GEM吸附磺酰化MSN包封微囊的GEM含有率为7.9w/w%(用10mMPBS的样品)以及8.1w/w%(用模拟体液的样品)。西他滨吸附磺酰化MSN包封微囊的评价—释放特性对获得的西他滨吸附磺酰化MSN包封微囊,测定在10mMPBS(pH7.4,150mMNaCl)或模拟体液(SimulatedBodyFluid:SBF)中的37℃的吉西他滨的释放特性。应该指出,模拟体液根据T.Kokubo,H.Kushitani,S.Sakka,T.KitsugiandT.Yamamuro,"Solutionsabletoreproduceinvivosurface-structurechangesinbioactiveglass-ceramicA-W",J.Biomed.Mater.Res.,24,721-734(1990)中的记载进行制备。获得的结果如图32的图表所示。在10mMPBS和模拟体液中,获得了差异大的吉西他滨放出特性。可以认为,其受离子强度或离子种类的不同所造成的影响。吉西他滨吸附磺酰化MSN包封微囊的评价—对A549细胞的细胞摄取特性以及细胞杀伤效果:此外,使用下面的样品,对GEM吸附磺酰化MSN包封微囊的对A549细胞(人肺胞基底上皮腺癌细胞)的细胞摄取特性以及细胞杀伤效果按照以下的顺序进行评价。样品号1:GEM吸附磺酰化MSN包封微囊样品号2:磺酰化MSN包封微囊样品号3:GEM吸附磺酰化MSN样品号4:磺酰化MSN样品号5:空微囊样品号6:仅GEM即,将以上述顺序获得的GEM吸附磺酰化MSN包封微囊(样品号1)、作为其原料的磺酰化MSN包封微囊(实施例II-3A)(样品号2),以及作为其原料的空微囊(实施例II-1)(样品号5)分别用Cy3标识(标识阴离子性聚合物数的25%)后,分别添加到以RPMI-1640培养基中2.2×103细胞/孔的密度具有A549细胞的板的各孔中,37℃下培养。GEM吸附磺酰化MSN包封微囊(样品号1)的添加量设为吉西他滨在培养基中为0.5μg/mL浓度的量,磺酰化MSN包封微囊(样品号2)以及空微囊(样品号5)的添加量,以MSN包封微囊换算量,调整为和GEM吸附磺酰化MSN包封微囊(样品号1)同量。此外,作为比较,将作为各MSN包封微囊的材料使用的MSN按与实施例II-3A相同的顺序磺酰化(通过MPTS表面处理巯基化后氧化)的磺酰化MSN(样品号4),以及该MSN上吸附吉西他滨的GEM吸附磺酰化MSN(GEM含有量8.1w/w%)(样品号3),以及仅吉西他滨(样品号6),分别添加到细胞进行培养。GEM吸附磺酰化MSN(样品号3)以及仅吉西他滨(样品号6)以吉西他滨在培养基中为0.5μg/mL浓度的方式添加,磺酰化MSN(样品号4)的添加量,以MSN换算量,调整为和GEM吸附磺酰化MSN(样品号3)同量。对从培养开始起到经过48小时以及72小时的时间点的各MSN包封微囊样品(样品号1、2、5)的细胞摄取量按照以下顺序进行评价。即,将培养细胞的细胞质通过钙黄绿素(INVITROGEN社制Calcein,AM),将细胞核通过赫司特(INVITROGEN社制Hoechst33342)分别染色,使用InCellAnalyzer1000(GE医疗生物科学社制),激发波长550nm下观察细胞内存在的来自各MSN包封微囊的Cy3荧光量。获得的结果如图33(a)所示。另外,使用台盼蓝(INVITROGEN社制Trypanbluestain、0.4%)识别细胞未摄取的荧光物质,同样地观察细胞内摄取的来自各MSN包封微囊的Cy3荧光量。获得的结果如图33(b)所示。相对于空微囊(样品号5)几乎没有被细胞摄取,明白了GEM吸附磺酰化MSN包封微囊(样品号1)以及磺酰化MSN包封微囊(样品号2)都被细胞摄取。此外,从培养开始起到经过48小时以及72小时的时间点的各样品(样品号1~6)所产生的的细胞杀伤效果按照以下顺序进行评价。即,使用CellCountingKit-8(同仁化学社制),按照生产者的指示,向各孔中添加试药,再培养1小时后,计算存活细胞数。培养48小时后的结果如图34(a),培养72小时后的结果如图34(b)所示。证实了GEM吸附磺酰化MSN包封微囊(样品号1)中,GEM吸附磺酰化MSN(样品号3)以及仅吉西他滨(样品6)具有同等程度的强的细胞杀伤效果。另外磺酰化MSN(样品号4)中的弱的细胞杀伤效果得到证实,磺酰化MSN包封微囊(样品号2)以及空微囊(样品号5)中的细胞杀伤效果未得到证实。吉西他滨吸附磺酰化MSN包封微囊的评价—对C26细胞的细胞摄取特性以及细胞杀伤效果:此外,用C26细胞(大鼠成骨细胞样细胞)代替A549细胞,以下说明点以外,按照和上述相同的顺序评价细胞摄取特性以及细胞杀伤效果。GEM吸附磺酰化MSN包封微囊(样品号1)、GEM吸附磺酰化MSN(样品号3)以及仅吉西他滨(样品号6)的添加量设为以吉西他滨在培养基中为0.1μg/mL浓度的量,磺酰化MSN包封微囊(样品号2)以及空微囊(样品号5)的添加量以MSN包封微囊换算量,调整为和GEM吸附磺酰化MSN包封微囊(样品号1)同量,磺酰化MSN(样品号4)的添加量以MSN换算量,调整为和GEM吸附磺酰化MSN(样品号3)同量。作为细胞摄取的评价结果,在不存在台盼蓝下的全细胞内的Cy3荧光量的测试结果如图35(a),台盼蓝染色下的活细胞内的Cy3荧光量的测试结果如图35(b)所示。对于任何的MSN包封微囊的样品,与A549细胞的情况相比,C26细胞中细胞摄取量变少。据此,可以认为关于细胞摄取量,根据细胞种类具有差别。此外,作为细胞杀伤作用的评价结果,培养48小时后的结果如图36(a),培养72小时后的结果如图36(b)所示。和A549细胞的情况相同,对于C26细胞,GEM吸附磺酰化MSN包封微囊(样品号1)中,证实了GEM吸附磺酰化MSN(样品号3)以及仅吉西他滨(样品6)具有同等程度的强的细胞杀伤效果。另外磺酰化MSN(样品号4)中的弱的细胞杀伤效果得到证实,磺酰化MSN包封微囊(样品号2)以及空微囊(样品号5)中的细胞杀伤效果未得到证实。吉西他滨吸附磺酰化MSN包封微囊的评价—使用小鼠的肿瘤治疗效果:准备7组、每组5只的移植了人肺癌细胞株A549的BALB/c裸鼠(7周龄,各样品),向其中6组分别将上述样品1~6(组1~6)静脉内给药。各样品的给药量设为200μL,各样品的浓度对于GEM吸附磺酰化MSN包封微囊(样品号1)、GEM吸附磺酰化MSN(样品号3)以及仅吉西他滨(样品6),对应小鼠体重调整为吉西他滨5mg/kg(体重),磺酰化MSN包封微囊(样品号2)以及空微囊(样品号5)的添加量以MSN包封微囊换算量,调整为和GEM吸附磺酰化MSN包封微囊(样品号1)同量,磺酰化MSN(样品号4)的添加量以MSN换算量,调整为和GEM吸附磺酰化MSN(样品号3)同量。另外,剩下的1组给予200μLPBS(组7)。给药后经过28天,测定肿瘤体积。肿瘤体积,使用游标卡尺测定肿瘤的最大直径以及最小直径,通过以下的计算式算出。V=(a2×b)/2V:肿瘤体积,a:最大直径,b:最小直径肿瘤体积的测试结果如图37的图表所示。对于磺酰化MSN包封微囊给药组(组2)、GEM吸附磺酰化MSN给药组(组3)、磺酰化MSN给药组(组4)、以及空微囊给药组(组5)的任一组,和PBS给药组(组7)间没有显著差异,没有发现显著的抗肿瘤效果。另外,GEM单独给药组(组6)中,虽然肿瘤生长有所抑制,但还是与PBS给药组(组7)间没有显著的差异,没有发现显著的抗肿瘤效果。另一方面,GEM吸附磺酰化MSN包封微囊给药组(组1)中,相对于PBS给药组(组7)自不用说,和GEM单独给药组(组6)相比,肿瘤生长也有显著地抑制,证实了显著的抗肿瘤效果。由此,根据本发明的GEM吸附磺酰化MSN包封微囊(组1),明白了GEM被有效地传递到肿瘤部位,和GEM单剂相比,能够以低给药量发挥高的抗肿瘤效果。吉西他滨吸附磺酰化MSN包封微囊的评价—使用小鼠的血中滞留性以及肿瘤分布的评价准备4组、每组3只的移植了人肺癌细胞株A549的BALB/c裸鼠(7周龄,各样品)。向其中2组将以Cy5标识MSN后的GEM吸附磺酰化MSN包封微囊(样品号1)静脉内给药(组A1以及A2),向剩下的2组将以Cy5标识MSN后的GEM吸附磺酰化MSN(样品号3)静脉内给药(组B1以及B2)。各样品的给药量为200μL,各样品的浓度相应于小鼠体重调整为吉西他滨5mg/kg(体重)。给在药后24小时(组A1以及B1)以及72小时(组A2以及B2)的时间点杀死动物,通过与上述相同的方法,基于Cy5荧光量,测定GEM吸附磺酰化MSN包封微囊以及GEM吸附磺酰化MSN的血中浓度以及肿瘤中的浓度。GEM吸附磺酰化MSN包封微囊以及GEM吸附磺酰化MSN的血中浓度的测试结果如图38的图表所示。明白了GEM吸附磺酰化MSN包封微囊给药组(组A1以及A2)中,和GEM吸附磺酰化MSN给药组(组B1以及B2)相比较,在给药后24小时以及72小时的任何时间点血中浓度都远远高出,血中滞留性优异。此外,GEM吸附磺酰化MSN包封微囊以及GEM吸附磺酰化MSN的肿瘤中浓度的测试结果如图39的图表所示。GEM吸附磺酰化MSN包封微囊给药组(组A1以及A2)中,和GEM吸附磺酰化MSN给药组(组B1以及B2)相比较,在给药后24小时以及72小时的任何时间点肿瘤中浓度都远远高出,肿瘤聚集性优异[实施例群III]低水溶性物质包封微囊及其制备方法[实施例III-1]基于β-半乳糖苷酶包封交联微囊的靛蓝染料包封微囊的制备材料:使用由不带电亲水性嵌段的聚乙二醇(PEG;分子量约2000)和阴离子性嵌段的聚天冬氨酸(P(Asp);聚合度约75)形成的阴离子性嵌段聚合物(PEG-P(Asp);Zeta电位-30.6mV)作为第1聚合物。使用由阳离子性嵌段的聚(含有两个氨基结构的天冬酰胺衍生物)(P(Asp-AP);聚合度约82)形成的阳离子性均聚物(Homo-P(Asp-AP);Zeta电位+16.3mV)作为第2聚合物。使用β-半乳糖苷酶作为被包封物质的酶酶包封微囊的调制及评价:将第1以及第2聚合物分别溶解在10mM磷酸缓冲液(pH7.4)(水性介质)中,调制各聚合物浓度1.0mg/mL的溶液。另外,将β-半乳糖苷酶(酶)以浓度1mg/mL的方式溶解在10mM磷酸缓冲液(pH7.4,含有150mM氯化钠)中,调制溶液。将获得的第1聚合物溶液以及第2聚合物溶液以电荷比相等(即C/A比=1.0)的方式加入到微量离心管混合,通过用涡旋搅拌器、2000rpm,搅拌2分钟,获得含有第1以及第2聚合物自组装而形成的微囊(空微囊)的溶液。向上述获得的微囊含有液中添加和使用的第2聚合物的溶液相同体积的上述β-半乳糖苷酶溶液,通过用涡旋搅拌器、2000rpm,搅拌2分钟,获得含有β-半乳糖苷酶包封微囊(酶包封微囊)的溶液。将获得的β-半乳糖苷酶包封微囊含有液添加到含有相对于PEG-P(Asp)有的羧基10倍等量EDC(同仁化学研究所制,WSC)的溶液中,静置一晚,通过EDC反应进行交联,通过离心超滤(VIVASPIN20、sartoriusstediumbiotech社制,截止分子量30万;2000rpm、25℃)将反应的副产物除去精制,获得含有交联的β-半乳糖苷酶包封微囊的溶液。对获得的β-半乳糖苷酶包封交联微囊含有液进行DLS测定,求出粒径分布、平均粒径以及PDI。粒径分布图如图40所示。观察到平均粒径为112nm的单分散粒子的形成。PDI为0.067。低水溶性物质包封微囊的调制及评价:将X-gal(5-溴-4-氯-3-吲哚基-β-D-吡喃半乳糖苷:水溶性前体),以浓度为5mg/mL的方式溶解在10mM磷酸缓冲液(pH7.4,含有150mM氯化钠)/二甲基甲酰胺混合溶液(4:1)中,调制溶液(应该指出,X-gal通过β-半乳糖苷酶的酶作用转变为靛蓝染料(5,5'-二溴-4,4'-二氯-靛蓝))。向50μL总聚合物浓度0.5mg/mL的上述β-半乳糖苷酶包封微囊含有液中添加10μL上述的X-gal溶液,而且加入190μL的10mM磷酸缓冲液,37℃下放置24小时,进行通过β-半乳糖苷酶(酶)的由X-gal(水溶性前体)向靛蓝染料(5,5'-二溴-4,4'-二氯-靛蓝:低水溶性物质)转变的反应。其后,用0.45μm的PES过滤器过滤,并且通过离心超滤(VIVASPIN20、sartoriusstediumbiotech社制,截止分子量使用30万;2000rpm、25℃)进行精制,获得含有靛蓝染料包封微囊(实施例III-1的低水溶性物质包封微囊)的溶液。对获得的靛蓝染料包封微囊含有液进行DLS测定,求出粒径分布、平均粒径以及PDI。粒径分布图如图41所示。观察到平均粒径为140nm的单分散粒子。PDI为0.091。此外,对该靛蓝染料包封微囊含有液,通过透射电子显微镜(TEM)进行形态观察。通过TEM(日本电子制JEM-1400)获得的TEM照片如图42所示。根据图42,证实了通过β-半乳糖苷酶的由X-gal(水溶性前体)转变成的靛蓝染料(低水溶性物质)局限在微囊的内部或是膜内。另外,通过高分辨率的TEM(日本电子制JEM-2100F)获得的TEM照片如图43所示,根据图43,可以观察到被认为是来自靛蓝染料的衍射线。据此,证实了微囊内包封的靛蓝染料为晶体。此外,将该靛蓝染料包封微囊在含有90%二甲基亚砜的水溶液中放置一晚,通过吸光光度法算出生成的靛蓝染料的包封量。其结果为,证实了相对于总聚合物(第1以及第2聚合物)的总量的66.9%的靛蓝染料被包封在微囊内。[比较例III-1]通过游离β-半乳糖苷酶的靛蓝染料包封微囊的制备对根据实施例III-1相同的顺序调制的空微囊含有液,不混合添加β-半乳糖苷酶溶液,直接根据实施例III-1相同的顺序通过EDC反应进行交联,获得交联后空微囊含有液。对获得的空交联微囊含有液进行DLS测定,求出粒径分布、平均粒径以及PDI。粒径分布图如图44所示。证实了平均粒径为98nm的单分散粒子的形成。PDI为0.076。对50μL总聚合物浓度0.5mg/mL的上述交联空微囊含有液,添加12.5μL和实施例III-1相同调制的浓度为1mg/mL的β-半乳糖苷酶溶液,以及10μL和实施例III-1相同调制的浓度为1mg/mL的X-gal溶液,并加入177.5μL10nM磷酸缓冲液,37℃下放置24小时。其后,用0.45μm的PES过滤器过滤,通过离心超滤(VIVASPIN20、sartoriusstediumbiotech社制,截止分子量30万;2000rpm、25℃)进行精制,获得含有靛蓝染料生成后的空微囊(比较例III-1中,最初,空的微囊)的溶液。对获得靛蓝染料生成后的空微囊含有液进行DLS测定,求出粒径分布、平均粒径以及PDI。粒径分布图如图45所示。观察到平均粒径为104nm的粒子。PDI为0.131。此外,对该靛蓝染料生成后的空微囊含有液,通过透射电子显微镜(TEM)进行形态观察。通过TEM(日本电子制JEM-1400)获得的TEM照片如图46所示。根据图46,虽然通过β-半乳糖苷酶和X-gal的反应生成了低水溶性物质靛蓝染料,但是几乎没有观察到包封在微囊内的靛蓝染料。此外,将该靛蓝染料生成后的空微囊在含有90%二甲基亚砜的水溶液中放置一晚,通过吸光光度法算出生成的靛蓝染料的包封量。其结果为,证实了相对于总聚合物(第1以及第2聚合物)的重量的31.0%的靛蓝染料粘附在微囊上。考虑到图46的结果,可以认为粘附在微囊上的靛蓝染料,不是包封在微囊内,主要是粘附在空微囊的外部。产业上使用的可能性根据本发明获得的物质包封微囊在用于药物传递的DDS,或生物材料、功能材料等领域中具有极高的可用性。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1