一种贵金属负载型催化剂的制备方法及催化剂和环烷烃氢解开环方法与流程
本发明涉及一种贵金属负载型催化剂和制备方法及应用以及使用该催化剂催化环烷烃氢解开环方法。
背景技术:
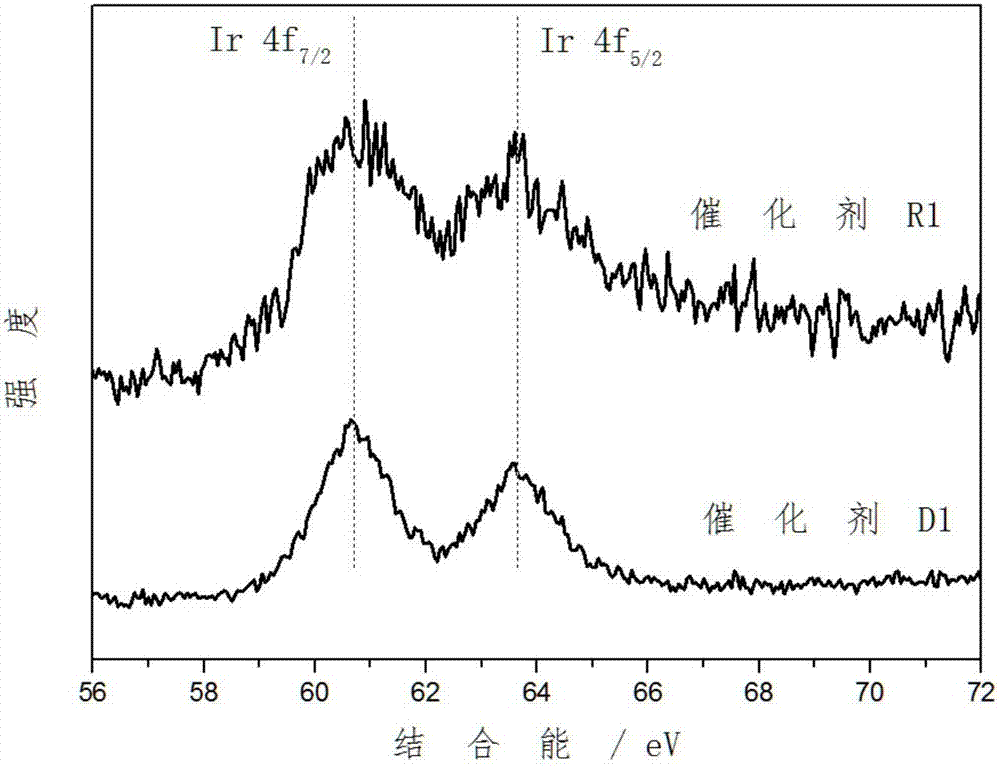
:随着世界经济的发展,柴油需求量日益增加。单靠直馏柴油不可能满足这一需求,这就需要调入二次加工柴油,如催化裂化柴油和焦化柴油。而二次加工柴油中含有大量的硫、氮和芳烃,目前硫和氮可以用传统的硫化物催化剂进行脱除,技术难点是芳烃的转化。柴油中高的芳烃含量不仅会降低油品质量,而且会增加柴油燃烧废气中的颗粒排放物。通常正构或短侧链烷烃的十六烷值最高,带有长侧链环烷烃及芳烃的十六烷值较高,而带有短侧链或无侧链环烷烃及芳烃的十六烷值最低。因此芳烃加氢饱和过程对提高柴油的十六烷值是有限的,而开环反应则有希望提高柴油的十六烷值。随着有关清洁能源的环境法规越来越苛刻,柴油燃料的脱芳改质成为研究的重点。因此,实现环烷烃的高选择性开环反应对于提高柴油品质有重要意义。环烷烃开环反应可以通过以下三个机理进行:自由基反应机理、正碳离子机理和氢解机理(journalofcatalysis,2002,210,137-148)。相比而言,金属催化的氢解机理对环烷烃选择开环反应具有较高的活性和选择性,主要是因为环烷烃分子的环内张力导致开环反应比断侧链反应容易进行。wo/2002/007881公开了一种用于环烷烃开环的催化剂和工艺,开环反应是通过使用氧化铝和酸性硅铝分子筛的复合载体负载的铱催化剂来实现的。而且,催化剂暴露于250℃氧气氛下焙烧再生,其开环活性不显著失活。cn200480043382.0公开了一种催化剂和使用该催化剂将环烷烃开环的方法。该催化剂包含第viii族金属组分、分子筛、难熔无机氧化物和非必要的改性剂组分。分子筛包括mapso、sapo、uzm-8和uzm-15,第viii族金属包括铂、钯和铑,而无机氧化物优选氧化铝。cn200910013536.6公开了一种环烷烃加氢转化催化剂及其制备方法和应用。催化剂包括载体和活性金属pt,载体由氢型y-beta复合分子筛和无机耐熔氧化物组成,催化剂载体中氢型y-beta复合分子筛含量为10wt%~90wt%,催化剂中活性金属pt的含量为0.05%~0.6%。催化剂采用浸渍法制备,得到的催化剂可以用于各种含环烷烃原料的加氢转化。cn201110102568.0公开了一种芳烃选择性开环反应工艺,反应在两个串联的反应器中进行;物料进入第一反应器进行深度脱硫和脱氮反应,经过h2s和nh3分离装置脱硫和氮,当物料中的s含量低于50ppm,n含量低于10ppm后,物料进入第二反应器进行选择性开环反应,该反应器有两个反应床层,在第一个反应床层进行加氢饱和异构化反应,第二个反应床层进行选择性开环反应;第一反应器选用金属硫化物催化剂;第二反应器的第一个床层装填贵金属/分子筛-氧化铝催化剂。醇热还原法过程使用醇(一般为多元醇)为还原剂和溶剂,而且大多不使用稳定剂,制备的催化剂颗粒分散度高、粒径分布均匀可控,且成本低廉、操作简单。wang报道了一种不使用稳定剂情况下,采用醇热还原法在有机介质中制备稳定的pt、rh、ru等单金属纳米粒子的方法(chemistryofmaterials,2000,12(6):1622-1627)。但是,上述公开的催化剂的环烷烃氢解开环活性和选择性仍有很大改善和提高的余地。上述公开的用于环烷烃氢解开环的贵金属负载型催化剂基本都是通过常规浸渍法或共浸渍法得到,然而,醇热还原法这一近年来所发展的新型制备高分散纳米金属催化剂的重要方法在适用于环烷烃氢解开环的催化剂领域鲜见报道。技术实现要素:本发明的目的在于提供一种贵金属负载型催化剂的制备方法及由该方法得到的催化剂及应用,还提供了一种催化环烷烃氢解开环反应的方法。本发明提供了一种贵金属负载型催化剂的制备方法,包括下述步骤:(1)配制含有含第viii族贵金属元素化合物的第一组分、含碱金属和\或碱土金属元素化合物的第二组分和含第vib和\或viib族金属元素化合物第三组分的混合溶液,在50-200℃条件下反应0.5-24小时,得到胶体溶液;(2)将载体分散于溶剂中,得到含载体的悬浊液;(3)将步骤(1)的胶体溶液与步骤(2)的悬浊液混合,然后经干燥和可选的焙烧,得到所述贵金属负载型催化剂。本发明还提供了由上述方法制得的贵金属负载型催化剂及其在催化环烷烃氢解开环反应中的应用。本发明进一步提供了一种环烷烃氢解开环方法,该方法包括在催化环烷烃氢解开环条件下,将含有环烷烃的原料、氢气与催化剂接触,其中,所述催化剂为上述贵金属负载型催化剂。与现有技术制备的相同贵金属含量的催化剂相比,本发明的贵金属负载型催化剂具有明显更高的催化环烷烃氢解开环活性,同时具有较低的裂解率。本发明的其它特征和优点将在随后的具体实施方式部分予以详细说明。附图说明:附图是用来提供对本发明的进一步理解,并且构成说明书的一部分,与下面的具体实施方式一起用于解释本发明,但并不构成对本发明的限制。在附图中:图1为本发明实施例1制得的催化剂r1和对比例1制得的对比催化剂d1的ir4f的x射线光电子能谱图;图2为本发明实施例1制得的催化剂r1和对比例1制得的对比催化剂d1的re4f的x射线光电子能谱图。具体实施方式以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。本发明提供了一种贵金属负载型催化剂的制备方法,包括下述步骤:(1)配制含有含第viii族贵金属元素化合物的第一组分、含碱金属或碱土金属元素化合物的第二组分和含第vib和/或viib族金属元素化合物第三组分的混合溶液,在50~200℃条件下反应0.5~24小时,优选在100-180℃条件下反应2-10小时得到胶体溶液;(2)将载体分散于溶剂中,得到含载体的悬浊液;(3)将步骤(1)的胶体溶液与步骤(2)的悬浊液混合,然后经干燥和可选的焙烧,得到所述贵金属负载型催化剂。其中,所述步骤(1)的三种组分可以根据各自溶解度、相溶性等性质,分开配制溶液后再混合,或直接在同一份溶液中配制;例如,分别配制含第一组分的溶液、以及同时含第二组分和第三组分的溶液,然后将两者混匀;或者是,分别配制同时含第一组分和第三组分的溶液、以及含第二组分的溶液,然后将两者混合。所述步骤(1)的含三种组分的溶液混合后,优选在惰性气氛和/或还原气氛下进行反应,所述惰性气氛和/或还原气氛可以是氮气、氩气、氦气、氢气以及它们的混合气。优选地,步骤(1)中,所述第viii族贵金属元素为选自ir、rh和ru中的至少一种,所述含第viii族贵金属元素化合物为选自h2ircl6、(nh4)2ircl6、ircl3、(nh4)3ircl6、rhcl3、(nh4)3rhcl6、rhpo4、rh2(so4)3、rucl3、(nh4)3rucl6、ru(ch3coo)3、ru(no)(no3)3中的至少一种;进一步优选地,所述第viii族贵金属元素为ir,所述含第viii族贵金属元素化合物为选自h2ircl6、(nh4)2ircl6、ircl3、(nh4)3ircl6中的至少一种。步骤(1)中所述碱金属或碱土金属元素优选自li、na、k、rb、ba中的至少一种,所述含碱金属或碱土金属元素的化合物优选为氢氧化物,所述第vib和/或viib族金属元素优选为mo、w、re、mn中的至少一种。优选地,步骤(1)中所述配制三种组分的溶液采用的溶剂与步骤(2)中所述溶剂可以是同样的溶剂,也可以是不同的溶剂,二者可以分别独立的选自醇或醇与水的混合物,所述醇为选自1-6个碳原子的一元醇、二元醇、三元醇中的至少一种,所述溶剂中醇的含量为40-100重量%;进一步优选地,所述醇为乙醇、乙二醇、1,2-丙二醇、1,3-丙二醇、丙三醇中的至少一种,所述溶剂中醇的含量为70-100重量%。为了最终催化剂性能更好,在制备胶体溶液时,优选地,步骤(1)中各组分的用量使得以所述胶体溶液重量为基准,以第viii族贵金属元素计的第一组分含量为1-200克/升,以碱金属和\或碱土金属元素计的第二组分含量为1-500克/升,以第vib和/或viib族金属元素计的第三组分含量为1-500克/升。所述步骤(2)的载体没有特别限定,优选地,可以为选自氧化铝、氧化硅、氧化钛、氧化镁、氧化锆、氧化钍、氧化铍、粘土、分子筛、活性炭中的一种或多种。所述步骤(2)的将载体分散于溶剂的方法没有特别限定,可以为本领域技术人员公知的各种方法;例如,可以为电磁搅拌分散、超声分散。在最终得到的催化剂中,优选地,步骤(1)和步骤(2)各组分的用量使得催化剂中以元素计的各组分含量如下:贵金属元素含量为0.2-15重量%,第vib和/或viib族金属元素含量为0.2-15重量%,碱金属和\或碱土金属含量为0-2%,其余为载体,满足总量为100重量%。进一步优选地,贵金属元素含量为0.5-10重量%,第vib和/或viib族金属元素含量为0.5-10重量%,碱金属和\或碱土金属含量为0-1%,其余为载体。步骤(1)的胶体溶液和步骤(2)的悬浊液混合后,可以先进行分离之后再干燥,也可以直接干燥。一般的,当固含量较低时,如50%以下时,优先采取分离后再干燥,固含量较高时,如50%以上时,直接干燥。所述步骤(3)的分离方法和干燥方式没有特别限定,可以为本领域技术人员公知的各种方法;分离方法可以采用常压过滤洗涤、减压过滤洗涤、离心分离洗涤,干燥方式可以是空气氛中烘箱干燥、真空干燥。对于干燥条件也没有特别限定,优选的干燥条件包括:温度40-200℃,时间0.1-24小时。还可以根据不同要求对上述干燥之后的产物进行焙烧或不焙烧,对焙烧的条件没有特别限制,优选地,可以是200-600℃下焙烧0.1-24小时。本发明还提供了由上述方法制得的贵金属负载型催化剂。优选条件下,以元素计并以催化剂总量为基准,贵金属含量为0.2-15重量%,第vib和/或viib族元素含量为0.2-15重量%,碱金属和/或碱土金属含量为0-2重量%,其余为载体;进一步优选地,贵金属含量为0.5-10重量%,第vib和/或viib族元素含量为0.5-10重量%,碱金属和/或碱土金属含量为0-1重量%,其余为载体。发明人对本发明得到的催化剂金属含量进行了测试,结果表明,本发明所述方法得到的贵金属负载型催化剂满足(m2/m1)xps/(m2/m1)xrf=2-20,优选2.5-10,更优选3-5,其中,m1是第viii族贵金属元素,m2是第vib和/或viib族金属元素,(m2/m1)xps是以x射线光电子能谱表征的催化剂中m2与m1以金属元素计的重量比,(m2/m1)xrf是以x射线荧光光谱表征的催化剂中m2与m1以金属元素计的重量比。所述x射线光电子能谱采用激发光源为150kw的单色器alkαx射线测得,所述x射线荧光光谱的测量条件包括铑靶、激光电压为50kv和激光电流为50ma。本发明还提供了上述贵金属负载型催化剂在催化环烷烃氢解开环反应中的应用。此外,本发明还提供了一种催化环烷烃氢解开环反应方法,该方法包括在催化环烷烃氢解开环条件下,将含有环烷烃的原料、氢气与催化剂接触,其中,所述催化剂为上述贵金属负载型催化剂。本发明的催化剂可用于各种含有环烷烃的原料的氢解开环反应,例如所述含有环烷烃的原料为环烷烃模型化合物,或者含环烷烃的汽油馏分、煤油馏分、或柴油馏分等,优选芳烃质量含量小于15%,硫质量含量小于30ppm。所述催化环烷烃氢解开环条件可以参照现有技术进行,优选情况下,温度为180-450℃,压力为1-18mpa,氢油体积比为50-10000:1,质量空速为0.1-100小时-1;进一步优选地:温度为220-400℃,压力为2-12mpa,氢油体积比为50-5000:1,质量空速为0.2-80小时-1。所述接触反应的装置可以在任何足以使所述原料油在加氢反应条件下与所述贵金属负载型催化剂接触反应的反应器中进行,例如固定床反应器、浆态床反应器、移动床反应器或沸腾床反应器。与现有技术制备的相同贵金属含量的催化剂相比,采用本发明的方法制得的催化剂具有明显更高的催化环烷烃氢解开环活性,同时具有较低的裂解率。究其原因,可能是因为本发明提供的催化剂的制备方法大幅度提高了贵金属与含第vib和/或viib族元素的物种的相互作用界面,有利于环烷烃中仲碳-叔碳键的选择性断裂,从而提高其开环选择性。以下的实施例便于更好地理解本发明,但并不限定本发明。下述实施例中,所述的百分含量,如无特别说明,均为质量百分含量;催化剂组成是以催化剂的总重量为基准,所述加氢活性金属元素的质量百分含量,且该组成根据投料量计算得到;x射线光电子能谱的测量仪器为thermoscientific公司的escalab250型仪器,测量条件为:激发光源为150kw的单色器alkαx射线,结合能采用c1s峰(284.8ev)校正;x射线荧光光谱的测量仪器为日本理学电机工业株式会社3271型仪器,测量条件为:粉末样品压片成型,铑靶,激光电压50kv,激光电流50ma。实施例1该实施例用于说明本发明提供的催化剂及其制备方法。配制100毫升含铱6.0克/升的氯铱酸和含铼10.0克/升的高铼酸的乙二醇溶液,在搅拌下将其加入到100毫升含钾18.5克/升的氢氧化钾的乙二醇溶液中,在室温下继续搅拌1小时,将所得反应物在氮气保护下于160℃下回流4小时,制得约200毫升的铱胶体溶液,冷却至室温备用。取20克sio2-al2o3载体(按照cn201110139331.x的实施例2制备,下同),电磁搅拌分散于50毫升乙醇中。在快速电磁搅拌下将上述200毫升的铱胶体溶液滴加到上述50毫升分散有载体的乙醇中,继续电磁搅拌2小时。将固体减压抽滤,水洗数次,于120℃真空干燥12小时,即制得含铱负载型催化剂,存于干燥器备用。得到的催化剂记为r1,其组成、xps和xrf表征结果见表1,其中x射线光电子能谱图如图1、图2所示。根据ir4f和re4f的电子结合能相应峰面积换算获得表层原子比值(m2/m1)xps。其中组成是以催化剂的总重量为基准,以元素计的所述铱、第三、第二组分金属元素的质量百分含量(下同)。对比例1该对比例用于说明对比催化剂及其制备方法。采用共浸渍法制备含铱负载型催化剂。配制44毫升含铱11.4克/升的氯铱酸和含铼15.9克/升的高铼酸的水溶液,等体积浸渍到20克sio2-al2o3载体,在20℃下搅匀,静置4小时后,经120℃烘干,在400℃焙烧4小时,400℃氢气还原4小时,氢气压力为0.1兆帕。还原后降至室温,存于干燥器备用。得到的催化剂记为d1,其组成、xps和xrf表征结果见表1。其中x射线光电子能谱图如图1、图2所示。对比例2该对比例用于说明对比催化剂及其制备方法。按照实施例1的方法制备催化剂,不同的是,制备铱胶体溶液所用的乙二醇溶液中不含高铼酸。得到的催化剂记为d2,其组成、xps和xrf表征结果见表1。实施例2该实施例用于说明本发明提供的催化剂及其制备方法。配制100毫升含铱6.0克/升的氯铱酸和含铼15.6克/升的高铼酸的乙二醇溶液,在搅拌下将其加入到100毫升含钾18.5克/升的氢氧化钾的乙二醇溶液中,在室温下继续搅拌1小时,将所得反应物在氮气保护下于160℃下回流4小时,制得约200毫升的铱胶体溶液,冷却至室温备用。取20克sio2-al2o3载体,电磁搅拌分散于50毫升乙醇中。在快速电磁搅拌下将上述200毫升的铱胶体溶液滴加到上述50毫升分散有载体的乙醇中,继续电磁搅拌2小时。将固体减压抽滤,水洗数次,于120℃真空干燥12小时,即制得含铱负载型催化剂,存于干燥器备用。得到的催化剂记为r2,其组成、xps和xrf表征结果见表1。实施例3该实施例用于说明本发明提供的催化剂及其制备方法。配制100毫升含铱10.3克/升的氯化铱的乙二醇溶液,在搅拌下将其加入到100毫升含钾3.2克/升的氢氧化钾和含钨15.6克/升的钨酸铵的乙二醇溶液中,在室温下继续搅拌1小时,将所得反应物在氮气保护下于160℃下回流6小时,制得约200毫升的铱胶体溶液,冷却至室温备用。取20克sio2-al2o3载体,电磁搅拌分散于50毫升乙醇中。在快速电磁搅拌下将上述200毫升的铱胶体溶液滴加到上述50毫升分散有载体的乙醇中,继续电磁搅拌2小时。将固体减压抽滤,水洗数次,于140℃真空干燥10小时,即制得含铱负载型催化剂,存于干燥器备用。得到的催化剂记为r3,其组成、xps和xrf表征结果见表1。实施例4该实施例用于说明本发明提供的催化剂及其制备方法。配制100毫升含钌6.0克/升的氯化钌的1,2-丙二醇溶液,在搅拌下将其加入到100毫升含钠8.4克/升的氢氧化钠和含钼10.0克/升的钼酸铵的1,2-丙二醇溶液中,在室温下继续搅拌1小时,将所得反应物在氮气保护下于160℃下回流4小时,制得约200毫升的钌胶体溶液,冷却至室温备用。取20克γ-al2o3载体(长岭催化剂厂产品,粒度20-40目),电磁搅拌分散于50毫升乙醇中。在快速电磁搅拌下将上述200毫升的钌胶体溶液滴加到上述50毫升分散有载体的乙醇中,继续电磁搅拌4小时。将固体减压抽滤,水洗数次,于140℃真空干燥8小时,即制得含钌负载型催化剂,存于干燥器备用。得到的催化剂记为r4,其组成、xps和xrf表征结果见表1。实施例5-8这些实施例用于说明本发明提供的催化剂对模型化合物甲基环戊烷的催化氢解开环结果。按照下述步骤分别评价催化剂r1、r2、r3和r4。在连续流动固定床微反装置上对催化剂进行活性评价,原料油为模型化合物甲基环戊烷,催化剂装填量为0.5克,反应条件为:压力为3.0兆帕,原料油进量为0.2毫升/分钟,氢油体积比为1000,温度为260℃,反应3小时后取样进行在线气相色谱分析。反应开始前,先在260℃、3.0兆帕氢压、流速200毫升/分钟的氢气氛还原2小时。反应结果列于表2。对比例3-4这些对比例用于说明对比催化剂的氢解开环活性。按照与实施例5相同的方法和条件评价对比催化剂d1和d2。反应结果列于表2。表1表2实施例催化剂甲基环戊烷转化率(%)直链烷烃选择性(%)对比例3d14746对比例4d24137实施例5r16952实施例6r26753实施例7r36650实施例8r46449实施例9-12这些实施例说明本发明提供的催化剂处理油品时的氢解开环活性。按照下述步骤分别评价催化剂r1、r2、r3和r4。在30毫升加氢装置上,以深度加氢脱硫并芳烃部分饱和后的催化裂化柴油为反应原料(总芳烃含量9.5重量%,硫含量8.1ppm,十六烷值39.2),进行油品的开环活性评价。催化剂装填量20毫升,并用石英砂稀释到30毫升,粒度皆为20~40目。反应开始前,先在290℃、6.0兆帕氢压、流速200毫升/分钟的氢气氛还原4小时。然后,在温度、压力不变的情况下,将液体体积空速1.5小时-1,氢油体积比800的条件下对催化剂进行活性评价,反应稳定24小时后取样,分析生成柴油的十六烷值。评价结果见表3。对比例5-6该对比例用于说明对比催化剂处理油品时的开环活性。按照与实施例9相同的方法和条件分别评价对比催化剂d1、d2。反应结果列于表3。表3催化剂处理油品评价结果实施例催化剂十六烷值增加值对比例5d19.3对比例6d28.89r111.810r211.111r311.712r410.9这些实施例结果说明,本发明所提供的催化剂与现有技术制备的相同贵金属含量的催化剂相比,具有更好的环烷烃开环活性,对柴油十六烷值具有更大的提高幅度。以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合。为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1