一种利用5‑磷酸吡哆醛制备固定金属亲合材料的方法与流程
本发明属于分析化学技术领域,具体涉及一种将5-磷酸吡哆醛用于制备固定金属亲合材料的方法,及其在样品前处理中的应用。
背景技术:
蛋白质磷酸化是一种可逆的转录后修饰,在各种生理功能中都有重要的作用。目前,质谱技术由于其高灵敏度、高通量被广泛用于磷酸化多肽的研究中。然而由于磷酸化蛋白在生物样品里的含量低,并且磷酸化多肽在质谱中的离子化效率比非磷酸化多肽要低,使得我们难以对其进行直接分析。因此,在质谱分析前对样品中的磷酸化多肽进行富集显得十分重要。
固定金属亲合色谱(immobilizedmetalaffinitychromatography,imac)作为一种快速、简单易用和经济的纯化技术,近年来被研究者广泛应用于磷酸化肽的富集。其基本原理是将金属离子固定于一定的配体上,然后利用金属离子与磷酸根基团之间的配位作用来萃取磷酸化多肽。吸附到材料上的磷酸化多肽可以通过磷酸盐或者碱性溶液解吸下来。其中,研究最多的配体为亚氨基二乙酸(ida)、次氮基三乙酸(nta)和磷酸根基团等,且磷酸根基团修饰的多孔硅胶比传统的imac材料体现出更好的选择性。因此,在接下来的几年中,人们制备了一系列含磷酸根基团的材料,并将其用于复杂生物样品中磷酸化多肽的分离和富集,然而,在制备含磷酸根基团的材料的过程中,经常需要用到一些毒性较大的试剂,并且合成过程较为复杂。
技术实现要素:
为了克服现有技术中存在的不足,本发明引入一种新的配体—5-磷酸吡哆醛(pyridoxal5′-phosphate,plp),它是维生素b6在体内的活性型代谢产物,且具有双功能反应基团,分别是醛基和磷酸根。
本发明提供的技术方案具体如下:
一种固定金属亲合材料的制备方法,包括以下步骤:
(1)将活化过的纳米材料y分散在乙醇中,加入3-氨丙基三乙氧基硅烷进行搅拌,得到氨基修饰的纳米材料y,即y-nh2;所述的纳米材料y为纳米sio2、fe3o4@sio2纳米微球或碳纳米管;
(2)向步骤(1)得到的y-nh2中加入ph=7.4的磷酸缓冲液,然后加入5-磷酸吡哆醛和nabh3cn,室温反应,y-nh2表面被磷酸根修饰,将所得固体产物清洗后室温干燥,得到y-nh2-plp;
(3)向金属盐溶液中加入y-nh2-plp,室温震荡5-10h,所得固体产物依次用乙醇、水和丙酮清洗,60℃真空干燥,得到固定金属亲合材料,即y-nh2-plp-mn+。
所述步骤(1)中,纳米sio2或fe3o4@sio2纳米微球的活化方式为:将纳米sio2或fe3o4@sio2纳米微球分散在盐酸中室温静置0.5h以上;碳纳米管的活化方式为:将碳纳米管分散在浓硫酸、浓硝酸体积比为3:2的混合液中50℃搅拌20h以上。
所述步骤(1)中,纳米sio2或fe3o4@sio2纳米微球与3-氨丙基三乙氧基硅烷一起进行搅拌的温度为室温;碳纳米管与3-氨丙基三乙氧基硅烷(aptes)一起进行搅拌的温度为70℃。
所述步骤(2)中,固体产物的洗涤方式为:依次用乙醇、二次水和丙酮清洗。
步骤(2)中,所述的金属盐为硫酸钛、氯化镓、氯化铁中的一种。
一种固定金属亲合材料,由上述制备方法制备得到。
上述固定金属亲合材料在样品前处理中的应用。
本发明的原理如下:
本发明通过席夫碱反应在氨基化纳米材料(y)表面修饰磷酸根基团,得到y-nh2-plp;继而在磷酸根基团上修饰金属离子,得到y-nh2-plp-mn+固定金属亲和色谱(immobilizedmetalaffinitychromatography,imac)吸附剂,该吸附剂的螯合剂为5-磷酸吡哆醛。
本发明通过改变纳米材料y,分别选择二氧化硅(sio2),氧化碳纳米管(ocnt)和包硅磁颗粒(fe3o4@sio2)制备了三种不同基地材料的y-nh2-plp(y=sio2、ocnt或fe3o4@sio2)。
本发明具有以下优点和有益效果:
(1)本发明的修饰方法简单、省时,且不会破坏基底材料的形貌特征,而且该修饰对基底材料进行功能化,增加了基底材料的实用性。
(2)本发明制备的固定金属亲合材料y-nh2-plp-mn+可作为imac吸附剂,利用金属离子与磷酸根的亲和作用,应用于生物样品中磷酸化肽的选择性富集,成功地应用于标准多肽混合液、牛奶酶解液、人类血清样品、鼠脑裂解液中磷酸化多肽的萃取等领域。
附图说明
图1为y-nh2-plp-mn+(以fe3o4@sio2-nh2-plp-ti4+为例)制备的流程图。
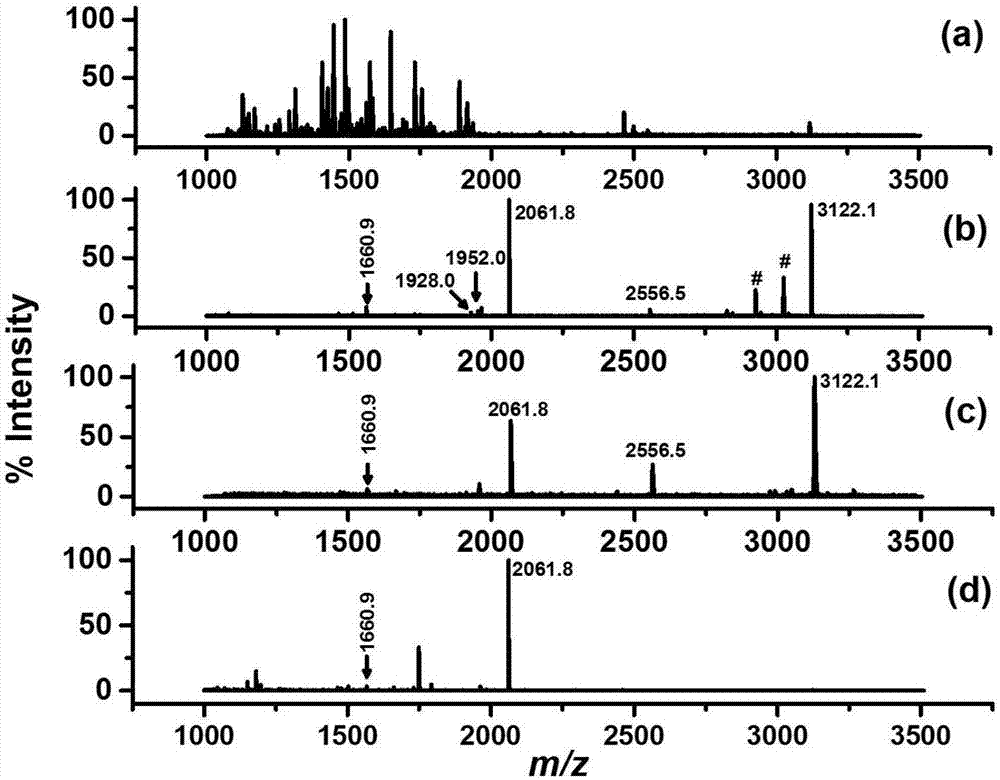
图2为β-酪蛋白和牛血清蛋白(bsa)酶解混合液(1:500)的质谱图;其中,图2(a)表示直接分析;图2(b)表示经过fe3o4@sio2-nh2-plp-ti4+萃取后;图2(c)表示经过sio2-nh2-plp-ti4+萃取后;图2(d)表示经过cnt-nh2-plp-ti4+萃取后;磷酸化多肽用其m/z值标记。
图3为β-酪蛋白和牛血清蛋白(bsa)酶解混合液(1:500)的质谱图;其中,图3(a)表示经过fe3o4@sio2-nh2-plp-ga3+萃取后;图3(b)表示经过fe3o4@sio2-nh2-plp-fe3+萃取后;磷酸化多肽用其m/z值标记。
图4为β-酪蛋白和bsa酶解混合液(1:1000)的质谱图;其中,图4(a)表示直接分析;图4(b)表示经过fe3o4@sio2-nh2-plp-ti4+萃取后;磷酸化多肽用其m/z值标记。
图5为血清和脱脂牛奶酶解液的质谱图;其中,图5(a)表示直接分析的血清样品;图5(b)表示fe3o4@sio2-nh2-plp-ti4+萃取后的血清样品;图5(c)表示直接分析的脱脂牛奶酶解液样品;图5(d)表示fe3o4@sio2-nh2-plp-ti4+萃取后的脱脂牛奶酶解液样品。
图6为鼠脑裂解液中磷酸化多肽的情况分析图。
具体实施方式
下面结合附图和实施例进一步阐述本发明,但这些实施例仅限于说明本发明,并不能限制本发明的范围。
实施例1
首先制备氨基修饰的纳米材料,得到y-nh2;再将5-磷酸吡哆醛接到y-nh2上,得到表面修饰磷酸根的y-nh2-plp;最后在磷酸根上固载金属离子,得到y-nh2-plp-mn+。
1.氨基修饰的纳米材料(y-nh2)的制备,根据纳米材料不同,其制备方法如下:
①sio2-nh2的制备:将0.5g纳米sio2分散在25ml1m的hcl水溶液中活化30min;经水及乙醇清洗后,将其分散在50ml乙醇中;在上述悬浮液中加入2mlaptes,室温下搅拌3h,得到氨基键合硅胶纳米颗粒。
②fe3o4@sio2-nh2的制备:将2.0gfecl2·4h2o和5.4gfecl3·6h2o溶解在2mhcl水溶液中,通入氮气除去溶解氧,然后加入30ml浓氨水(25wt%),搅拌30min,得到fe3o4纳米颗粒;将fe3o4纳米颗粒经反复磁分离及水洗后,分散于50ml水中待用。
取5mlfe3o4纳米颗粒悬浮液,用乙醇清洗后,分散于40ml乙醇中,并超声1h;之后加入1.5ml浓氨水,6ml水以及1.5mlteos,并在40℃下搅拌2h。最后所得的fe3o4@sio2纳米颗粒经去离子水及乙醇清洗后,置于60℃真空干燥箱中烘干待用。将0.5gfe3o4@sio2分散在25ml1m的hcl水溶液中活化30min;经水及乙醇清洗后,将其分散在50ml乙醇中;在上述悬浮液中加入2mlaptes,并在室温下搅拌3h,得到氨基键合硅胶纳米颗粒。
③ocnt-nh2的制备:将碳纳米管(cnt)分散于300ml浓硫酸、浓硝酸体积比为3:2的混合液中,在50℃搅拌20h,然后将所得溶液过滤,并用水和丙酮清洗,在100℃真空条件下干燥24h,将所得产物氧化碳纳米管(ocnt)分散于50ml乙醇中,超声30min。随后加入2mlaptes于70℃搅拌4h,得到氨基键合的氧化碳纳米管,即ocnt-nh2。
2.y-nh2-plp的制备:称取0.4gy-nh2于50ml圆底烧瓶中,加入40ml0.1m的磷酸缓冲液(ph7.4),再加入0.2gplp和0.2g氰基硼氢化钠(nabh3cn),25℃反应5h。产物依次用乙醇、二次水和丙酮清洗,室温干燥过夜,即得到y-nh2-plp。
3.y-nh2-plp-mn+的制备:配制三份硫酸钛水溶液(0.1m)20ml,分别加入0.2gy-nh2-plp;分别配制氯化镓水溶液(0.1m)、氯化铁水溶液(0.1m)各20ml,分别向这两份溶液中加入0.2gfe3o4@sio2-nh2-plp,室温震荡5h,产物依次用乙醇、水和丙酮清洗,60℃真空干燥,得到固定金属亲合材料,即y-nh2-plp-mn+。
实施例2
为了考察三种不同纳米材料修饰钛离子(y-nh2-plp-ti4+)对磷酸化肽的萃取效果,从而选出一种最优的基底材料,我们比较了三种y-nh2-plp-ti4+(y=sio2、ocnt或fe3o4@sio2)在β-酪蛋白和bsa酶解混合液中对磷酸化多肽的萃取效果:
将β-酪蛋白用100mmtris-hcl(ph=8.5)缓冲液配制成浓度为1mg/ml的溶液;在上述溶液中按照酶与底物比为1:50(w/w)的比例加入胰蛋白酶,并在37℃反应16h。酶解后的多肽产物经真空离心浓缩仪抽干后存放在-20℃的冰箱中备用。
称取1mgbsa溶于100μl含8m尿素及100mmtris-hcl的变性缓冲液(ph=8.5)中,然后加入5μl100mm三(2-甲酰乙基)膦盐酸盐溶液(三羧甲基磷酸,tcep),并在室温下反应20min以还原蛋白质中的二硫键,继续加入5μl100mm碘乙酰胺(iaa)溶液,并在避光条件下,室温下反应30min使被还原的巯基烷基化。经还原及烷基化的蛋白质用300μl100mmtris-hcl(ph8.5)稀释后,按照酶与底物比为1:50(w/w)的比例加入胰蛋白酶,并在37℃反应16h。酶解后的多肽产物抽干后存放在-20℃的冰箱中备用。
为了比较基于三种不同基底材料y-nh2-plp-ti4+(y=sio2、ocnt或fe3o4@sio2)对磷酸化肽段的富集能力,将磷酸化蛋白(β-酪蛋白)与非磷酸蛋白(bsa)酶解产物的混合物作为分析物(β-酪蛋白:bsa=1:500)。
分别称取10mgy-nh2-plp-ti4+分散于1ml水中,混合均匀后,取20μl分散液,用上样液(5%tfa-50%acn(v/v))平衡,然后将其加入100μl含多肽混合物的上样液中,涡旋3min,离心或磁分离,经过上样液清洗两遍之后,材料上的分析物用100μl解吸液(0.4mnh3·h2o)解吸到离心管中。解吸液经真空离心浓缩仪抽干后,加入1.2μl基质溶液(20mg/ml2,5-dhb于50%acn,1%h3po4的水溶液中)溶解多肽,全部样品点在maldi靶上用于maldi-tofms分析。
所有的质谱分析都采用日本shimadzu公司的aximatof2型基质辅助激光解吸电离-飞行时间质谱仪(maldi-tofms)。maldims分析时采用波长为337nm的氮气激光器,其脉冲宽度为3ns;采用20kv的加速电压,在正离子反射模式下进行检测。每张谱图是200次激光扫描的平均结果。
检测结果如图2所示:当β-酪蛋白酶解物未经富集直接经maldims分析时,谱图中存在大量非磷酸化多肽的峰,没有磷酸化多肽的峰(图2(a))。经过三种y-nh2-plp-ti4+分别处理之后,解吸液中分别可检测到6、4、2个磷酸化多肽的信号峰(图2(b),图2(c),图2(d)),而绝大多数非磷酸化多肽的信号被有效地消除,表明这三种y-nh2-plp-ti4+对磷酸化多肽均表现出一定的选择性。从质谱图中磷酸化多肽个数和杂峰情况来看,以fe3o4@sio2为基底的材料富集效果最好,所能检测到的磷酸化多肽的信息如表1所示。推测的原因是,相比于磁纳米颗粒,硅胶颗粒粒径更大,分散性较差,吸附量较小,且分离需要离心,操作不方便;而碳纳米管表面具有一定的疏水性,会增加对非磷酸化多肽的非特异性吸附。
实施例3
为了考察三种常用金属离子(ti4+,ga3+,fe3+)对磷酸化肽的萃取效果,从而选出一种最优的金属离子以获得最优的材料,我们比较了fe3o4@sio2-nh2-plp-mn+(m=ti4+、ga3+或fe3+)在β-酪蛋白和bsa酶解混合液中对磷酸化多肽的萃取效果:
为了比较三种键合不同金属离子的fe3o4@sio2-nh2-plp-mn+(m=ti4+、ga3+或fe3+)对磷酸化肽段的富集能力,将磷酸化蛋白(β-酪蛋白)与非磷酸蛋白(bsa)酶解产物的混合物作为分析物(β-酪蛋白:bsa=1:500)。
分别称取10mgfe3o4@sio2-nh2-plp-mn+分散于1ml水中,混合均匀后,取20μl分散液,用上样液(5%tfa-50%acn(v/v))平衡,然后将其加入100μl含多肽混合物的上样液中,涡旋3min,离心或磁分离,经过上样液清洗两遍之后,材料上的分析物用100μl解吸液(0.4mnh3·h2o)解吸到离心管中。解吸液经真空离心浓缩仪抽干后,加入1.2μl基质溶液(20mg/ml2,5-dhb于50%acn,1%h3po4的水溶液中)溶解多肽,全部样品点在maldi靶上用于maldi-tofms分析。
检测结果如图3所示:fe3o4@sio2-nh2-plp-fe3+和fe3o4@sio2-nh2-plp-ga3+这两种材料对磷酸化多肽也表现出一定的选择性,尤其是多磷酸化多肽。从质谱图中磷酸化多肽个数和杂峰情况来看,fe3o4@sio2-nh2-plp-ti4+表现得最好(图2(b))。推测原因是,fe3+和ga3+相比于ti4+与磷酸化多肽的相互作用力更弱,因此损失大量的单磷酸化多肽。
实施例4
为了进一步评价fe3o4@sio2-nh2-plp-ti4+对磷酸化多肽的萃取能力,我们将fe3o4@sio2-nh2-plp-ti4+应用于更复杂的β-酪蛋白和bsa酶解混合液中磷酸化多肽的萃取,同时继续提高磷酸化蛋白(β-酪蛋白)与非磷酸蛋白(bsa)酶解产物的混合物中bsa的比例。
称取10mgfe3o4@sio2-nh2-plp-ti4+分散于1ml水中,混合均匀后,取20μl分散液,用上样液(5%tfa-50%acn(v/v))平衡,然后将其加入100μl含多肽混合物的上样液中,涡旋3min,离心或磁分离,经过上样液清洗两遍之后,材料上的分析物用100μl解吸液(0.4mnh3·h2o)解吸到离心管中。解吸液经真空离心浓缩仪抽干后,加入1.2μl基质溶液(20mg/ml2,5-dhb于50%acn,1%h3po4的水溶液中)溶解多肽,全部样品点在maldi靶上用于maldi-tofms分析。
检测结果如图4所示:当β-酪蛋白酶解物和bsa酶解物的比例为1:1000时,在未经过前处理的谱图中只能看到大量的非磷酸化肽信号(图4(a));经过fe3o4@sio2-nh2-plp-ti4+处理之后,谱图中可以看到6个磷酸化肽的峰,而且大量的非磷酸化肽信号被去除(图4(b)),说明fe3o4@sio2-nh2-plp-ti4+对磷酸化肽有极好的选择性。
实施例5:fe3o4@sio2-nh2-plp-ti4+应用于血清中内源性磷酸化多肽的富集
正常人的血清样品通过标准的临床渠道从武汉大学校医院获得,采集到的血清保存在-20℃冰箱中。
称取10mgfe3o4@sio2-nh2-plp-ti4+分散于1ml水中。混合均匀后,取20μl,用上样液(5%tfa-50%acn(v/v))平衡后,进行萃取。萃取磷酸化多肽时,血清(2μl)需要用上样液稀释50倍再进行萃取。上样液和解吸液分别为100μl5%tfa-50%acn(v/v)和0.4m氨水水溶液。萃取完成之后,解吸液进行冷冻旋干,1.2μl基质溶液(20mg/ml2,5-dhbin50%(v/v)acn,1%(v/v)h3po4)溶解多肽,全部样品点在maldi靶上用于maldi-tofms分析。
检测结果如图5所示:血清未经过材料处理直接进样时,质谱图中只能非磷酸多肽的信号(图5(a));经过材料处理之后,质谱图中清晰的呈现着4个磷酸化多肽,而且几乎看不到非磷酸化肽的信号(图5(b),附表2),表明材料适用于高蛋白生物样品的分析。
实施例6:fe3o4@sio2-nh2-plp-ti4+应用于脱脂牛奶酶解物中磷酸化多肽的选择性富集
脱脂牛奶购买自武汉大学某超市。牛奶的酶解过程如下:取100μl低脂牛奶经真空离心浓缩仪抽干后复溶于100μl含8m尿素及100mmtirs-hcl的变性缓冲液(ph8.5)中。上述溶液经10mm二硫苏糖醇(dtt)于37℃下反应30min以还原蛋白质中的二硫键,之后在20mm碘乙酰胺(iaa)中于避光条件下,室温下反应30min使被还原的巯基烷基化。经还原及烷基化的蛋白质用300μl100mmtris-hcl(ph8.5)稀释后,按照酶与底物比为1:50(w/w)的比例加入胰蛋白酶,并在37℃反应16h。所有的酶解物保存在-20℃冰箱中备用。酶解后的多肽样品稀释500倍后用于材料的选择性评价。
称取10mgfe3o4@sio2-nh2-plp-ti4+分散于1ml水中,混合均匀后,取20μl分散液,用上样液(5%tfa-50%acn(v/v))平衡后,进行萃取。萃取磷酸化多肽时,牛奶酶解物用上样液稀释500倍再进行萃取。上样液和解吸液分别为100μl5%tfa-50%acn(v/v)和0.4m氨水水溶液。萃取完成之后,解吸液进行冷冻旋干,1.2μl基质溶液(其中,2,5-dhb浓度为20mg/ml,50%(v/v)acn,1%(v/v)h3po4)溶解多肽,全部样品点在maldi靶上用于maldi-tofms分析。
检测结果如图5所示:牛奶酶解物未经过材料处理直接进样时,质谱图中只能看到7个磷酸化多肽以及大量的非磷酸化肽(图5(c));经过材料处理之后,质谱图中清晰的呈现着20个磷酸化多肽(图5(d),附表3),表明材料可用于实际样品分析。
实施例7:fe3o4@sio2-nh2-plp-ti4+应用于鼠脑裂解液中磷酸化多肽的富集
斯普拉格(sd)雌性大鼠由湖北省疾控中心提供,年龄3~4个月,体重250~300g。鼠脑被取下后,清洗干净,切碎。取0.5g鼠脑组织,用匀浆管研碎,加入1mlripa裂解液(含磷酸酶抑制剂和蛋白酶抑制剂)。在冰浴中裂解30分钟后,于4℃下12000rpm离心30min,收集上清液。利用bca方法对上述上清液中的蛋白进行定量。往上述上清液中加入3体积的丙酮/乙醇/乙酸混合液(50/50/0.1,v/v/v)进行蛋白沉淀。之后4℃下4000rpm离心30min;去除上清液后,得到蛋白质,在空气中晾干。蛋白质用含8m尿素和0.2mtris、4mmcacl2的溶液复溶后同上述步骤进行酶解。酶解后的多肽产物经c18柱脱盐并抽干后存放在-20℃的冰箱中备用。
称取10mgfe3o4@sio2-nh2-plp-ti4+分散于1ml水中,混合均匀后,取20μl分散液,用上样液(5%tfa-50%acn(v/v))平衡后,进行萃取。萃取鼠脑裂解液样品时,鼠脑蛋白的用量为0.5mg。上样液和解吸液分别为100μl5%tfa-50%acn(v/v)和0.4m氨水水溶液。解吸液经真空离心浓缩仪抽干后,用c18柱spe脱盐,然后用反相液相色谱-电喷雾串联质谱(lc-esi-ms/ms)分析。
经富集后的细胞裂解物酶解产物采用nanorplc-esi-ms/ms(tripletof5600+,absciex)进行质谱分析。多肽首先被结合到captrap柱(5μm,5×0.3mm,agilenttechnologies)上,然后被洗脱到反相c18分析柱(75μm×150mm,3μmparticlesize,
检测结果如图6所示:该方法在0.5mg鼠脑裂解液中共检测到3014个磷酸化多肽,其中单磷酸化多肽有2848个,多磷酸化多肽有166个,对磷酸化多肽的特异性为88.1%(图6)。结果表明该材料在复杂生物样品的分析中表现良好,有望用于磷酸化蛋白组的全分析。
表1β-酪蛋白酶解液中鉴定到的磷酸化多肽的详情
“_”表示磷酸化位点
表2人类血清中鉴定到的磷酸化多肽的详情
“_”表示磷酸化位点。
表3脱脂牛奶酶解液中鉴定到的磷酸化多肽的详情
“_”表示磷酸化位点。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!