一种无金属H2S选择性催化氧化催化剂及制备方法与流程
本发明涉及一种以糖类为原料的多孔纳米碳材料制备方法,具体涉及一种新型无金属h2s选择性氧化催化剂的制备及其应用方法,该方法可同时实现h2s气体的去除和硫磺的回收再利用。
背景技术
h2s作为大气的主要污染物之一,是一种高刺激性剧毒气体,在有氧和湿热条件下,不仅会引起设备和管路腐蚀、催化剂中毒,而且会严重威胁人身安全。随着经济的发展,人们环保意识的增强,尾气的脱硫问题越来越受到人们的关注。同时国家也制定了相应的法律、法规,对h2s排放量作了严格的限制。硫化氢是许多工业生产中的副产物。目前,有70多种职业会接触到硫化氢。这些职业包括采矿、石油开采与提炼、皮革制造、橡胶合成、煤气制取、人造纤维、造纸、染料、印染、制糖、食品加工等。此外,有机物腐败场地也有硫化氢产生,并易积聚在通风不良的城市污水管道、窖井、化粪池、污水池、纸浆池以及其他各类发酵池和蔬菜腌制池等封闭和半封闭的设施、容器及管道内或低洼处、因此清理这些场所时,也会接触硫化氢。另外,硫化氢可溶于水和甲醇、乙醇,与空气混合能形成爆炸性混合物;遇明火、高温能引起燃烧爆炸。目前,在工业上h2s的脱除主要采用克劳斯(claus法)工艺。该工艺需先吸收h2s后提浓,再通过进一步催化处理制取单质硫。能达到脱除h2s气体的目的,又可实现其中硫元素的回收利用。但是由于热力学限制,克劳斯尾气中还含有3%-5%的硫化物未能转化成单质硫。随着环境法规日益严格,需要寻找一种不受热力学平衡限制,可高效脱除h2s并实现硫单质回收利用的处理方法。
近年来,选择性催化氧化h2s的方法受到人们的广泛重视,反应如式(1)所示。h2s选择性催化氧化法不受热力学平衡限制,理论h2s转化率可以达到100%。并且该反应工艺先进,过程简单。因反应是放热反应,h2s含量0.3%以上即可不需要外来热量,能耗低。因此,该反应具有良好的应用前景,实现这一过程的关键在于开发具有高效的催化活性及选择性的催化剂。
h2s+1/2o2→(1/n)sn+h2o(1)
目前应用于h2s选择性催化氧化领域的催化剂主要有传统碳材料、分子筛和金属氧化物。但从文献上看,以上催化剂仍存在不足之处。例如,活性炭、分子筛材料因其比表面积大、孔道丰富使其传质过程及产物硫脱附速率加快,但是该材料自身催化中心少,需要负载活性组分或进行改性后才具有催化性能,因而存在制备过程繁琐和活性组分在反应中易流失等问题;碳纳米管具有独特的一维结构,且石墨化程度较高,一定湿度下,h2s解离的hs-离子易于在其表面快速迁移,具有较好的氧化脱硫性能;但受限其比表面积小,饱和硫容最高仅达到1.86gh2s/g催化剂。介孔碳具有较大的中孔孔隙,有利于硫的存储;同时通过氮掺杂可提高材料的表面碱性,增加碳边缘位和缺陷位的数量,强化h2s的吸附解离,增强其催化能力。氮掺杂介孔碳的穿透硫容可提高至2.77gh2s/g催化剂。但介孔碳材料的制备方法复杂,合成技术要求高,不易实现工业化生产。金属氧化物自身有催化活性位点,而且稳定性比较高,但比表面积较小,限制了其对h2s的吸附,而且在反应中容易发生硫磺覆盖活性中心,使催化剂性能大幅降低。因此,除了在原有载体上改性外,发展新型高效的h2s选择性催化氧化催化剂是目前实现硫化氢脱除技术突破的必经之路。
经研究发现,具有丰富的孔道结构、大的比表面积和大量均匀分散的活性位点的催化剂有利于h2s的吸附和氧化还原反应的发生。鉴于传统的碳材料等催化剂存在比表面积小和活性位点少等许多的不足,我们提出一种简便的多糖系列碳材料的制备方法,并且与其他文献和专利报道的类似材料相比,我们提出的制备方法绿色环保,操作方法简单,而且制备的催化剂具有高的硫容、高的比表面积、丰富的孔道结构和活性位点等特点,这也是将该材料应用于硫化氢选择性催化氧化反应的关键。
技术实现要素:
本发明的目的在于解决现有h2s选择性催化氧化催化剂存在的问题,致力于开发一种制备方法简便,催化性能优异的催化剂。通过简单绿色的机械研磨将原料均匀混合,在惰性气氛中的经过高温焙烧处理后,得到一种高效h2s选择性催化氧化无金属催化剂。通过调控球磨机的转速和焙烧温度来获得不同氮含量及粒径孔径的多孔碳材料。该方法所制备的多孔碳材料催化剂在h2s选择性催化氧化中显示出优异的催化性能。该发明也是首次将这种方法制备的多孔碳材料用于h2s选择性催化氧化反应中,同时为开发绿色高效,以及方法简便的h2s选择性催化氧化催化剂提供了新思路。
为了实现上述设计催化剂的目的,本发明将采用如下的技术方案:
一种无金属h2s选择性催化氧化催化剂的制备,以糖类为主要的碳源,以双氰胺等为氮源,机械研磨先经过研钵的研磨,筛分至一定的目数,然后在球磨机中混合均匀,球磨得到微米级的混合物,经过在惰性气体中高温焙烧得到样品。
其具体步骤如下:
(1)称取一定量的葡萄糖、壳聚糖、蔗糖等糖类中的至少一种作为碳源,将其置于研钵中并充分研细,经过标准筛筛分后得到120~140目的原料a;
(2)称取一定量的双氰胺、三聚氰胺、尿素等药品中的至少一种作为氮源,将其置于研钵中并充分研细,经过标准筛筛分后得到120~140目的原料b;
(3)将得到的原料a、b按一定的质量比例(a:b=0.02~0.1:1)混合,混合均匀后装入100ml的球磨罐中,玛瑙磨球按大球占20%、中球占50%、小球占30%的质量比例装入球磨罐中,以180~240转/分的转速,按顺时针方向球磨30~60分钟,然后再按逆时针方向球磨30~60分钟;
(4)待球磨结束后,将球磨罐里的混合物置于陶瓷方舟中,并放入石英管式炉中,接着开启真空泵抽30~60分钟,然后将氮气通入管式炉中,并由室温经过4~6小时程序升温到400~600℃,接着经过1~4小时程序升温到最终碳化温度(700~900℃)进行高温碳化1~4小时,待冷却到室温收集得到催化剂,并可用于h2s选择性催化氧化反应中。
无金属催化剂的应用:将其应用于选择性催化氧化h2s为硫单质和水的催化反应中,反应温度为100℃~200℃,原料气为5000ppmh2s,2500ppmo2,n2为平衡气的三组分气体,原料气流速v为20ml·min-1,反应管内径为5mm。
步骤中所述的催化剂使用于h2s选择性催化氧化反应上,其性能评价公式如下:
由于目前h2s选择性催化氧化存在许多的问题,例如制备方法繁琐复杂难以工业化,催化剂容易永久失活等。为此本发明提供了一种h2s选择性催化氧化催化剂的简单制备方法,以糖类为最主要的碳源,双氰胺、脲和三聚氰胺等为主要的氮源,经过简单的筛分和机械球磨混合,在惰性气氛的高温管式炉中焙烧即可得到催化剂。
本发明的上述技术方案相比现有技术具有以下优点:
(1)本发明所采用的无金属催化剂制备方法简单,选用的合成原材料易得,成本低,绿色无污染,有利于工业化的大规模生产,并且具有普适性,应用前景广阔;
(2)本发明无金属h2s选择性催化氧化催化剂,具有大量的活性位点,丰富的孔道结构和较大的比表面积;
(3)本发明所述的无金属h2s选择性催化氧化催化剂无需负载或添加其他活性组分,在高转化率和选择性的同时,还具有较好的催化稳定性;
(4)本发明所述的无金属h2s选择性催化氧化催化剂,选择以绿色环保的糖类为主要碳源,双氰胺、脲和三聚氰胺的含氮量高的原料为主要的氮源,可以降低催化剂的生产成本,同时合成的催化剂能够获得15~45wt%的吡啶氮含量,目前的研究表明吡啶氮可以作为h2s选择性催化氧化过程中的活性位点,与普通的氮掺杂相比,含有较高含量吡啶氮的无金属催化剂催化性能得到了显著的提高;
(5)本发明所述的无金属h2s选择性催化氧化催化剂,采用慢速焙烧的制备方法,该方法所制备的催化剂与快速焙烧的方法相比较,能够产生更多的孔道结构,同时也增大了催化剂的比表面积。
附图说明
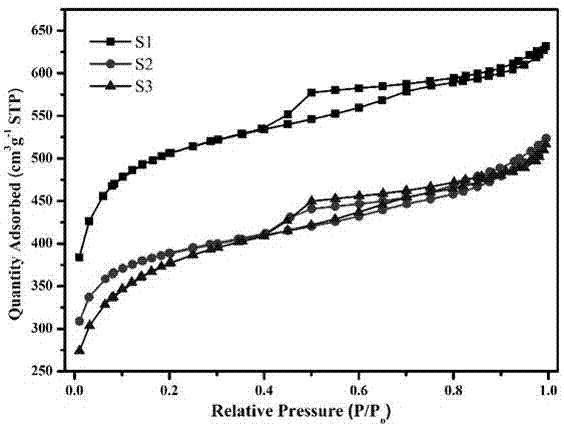
图1为实施例1~3所述样品的氮气吸脱附等温线;
图2为实施例1~3所述样品的孔径分布曲线;
图3为实施例1~3和对比例1~2所述样品进行h2s催化氧化催化性能评价中的h2s转化率随温度变化的曲线图;
图4为实施例1~3和对比例1~2所述样品进行h2s催化氧化催化性能评价中的s选择性随温度变化的曲线图;
图5为实施例1~3和对比例1~2所述样品进行h2s催化氧化催化性能评价中的硫产率随温度变化的曲线图;
图6为实施例1~3和对比例1~2所述样品,在180℃下反应25小时,h2s催化氧化催化稳定性能评价中的h2s转化率随反应时间变化的曲线图。
具体实施方式
为了使本发明的内容更容易被清楚的理解,下面根据本发明的具体实施例并结合附图,对本发明作进一步详细的说明。
实施例1:
取适量的葡萄糖于研钵中,研磨15~30分钟后,用标准筛筛分得到120目以下的葡萄糖,记为原料a;取适量的三聚氰胺于研钵中,研磨15~30分钟后,用标准筛筛分得到120目的三聚氰胺,记为原料b。用分析天平称取原料a为1g,称取原料b为35g,将原料a和b按1:35的质量比于研钵中充分混合,然后将混合均匀的混合物装入100ml的球磨罐中,玛瑙磨球按大球占20%、中球占50%、小球占30%的质量比例装入球磨罐中,以230转/分的转速,按顺时针方向球磨30分钟,然后按逆时针方向球磨30分钟。待球磨结束后,将球磨罐里的混合物置于陶瓷方舟中,并放入石英管式炉中,接着开启真空泵抽30分钟,然后将氮气通入管式炉中,由室温经过4小时程序升温到400℃,接着经过2小时程序升温到700℃进行焙烧2小时,待冷却到室温收集得到样品,记为催化剂s1,其氮含量为20~25wt%,粒径为2~5μm。
实施例2(最佳实施例):
取适量的壳聚糖于研钵中,研磨15~30分钟后,用标准筛筛分得到120目以下的壳聚糖,记为原料a;取适量的二氰二胺于研钵中,研磨15~30分钟后,用标准筛筛分得到130目的二氰二胺,记为原料b。用分析天平称取原料a为1g,称取原料b为35g,将原料a和b按1:35的质量比于研钵中充分混合,然后将混合均匀的混合物装入100ml的球磨罐中,玛瑙磨球按大球占20%、中球占50%、小球占30%的质量比例装入球磨罐中,以230转/分的转速,按顺时针方向球磨40分钟,然后按逆时针方向球磨40分钟。待球磨结束后,将球磨罐里的混合物置于陶瓷方舟中,并放入石英管式炉中,接着开启真空泵抽30分钟,然后将氮气通入管式炉中,由室温经过5小时程序升温到500℃,接着经过3小时程序升温到800℃进行焙烧2小时,待冷却到室温收集得到样品,记为催化剂s2,其氮含量为35~45wt%,粒径为500nm~1μm。
实施例3:
取适量的蔗糖于研钵中,研磨15~30分钟后,用标准筛筛分得到120目以下的蔗糖,记为原料a;取适量的尿素于研钵中,研磨15~30分钟后,用标准筛筛分得到140目的尿素,记为原料b。用分析天平称取原料a为1g,称取原料b为35g,将原料a和b按1:35的质量比于研钵中充分混合,然后将混合均匀的混合物装入100ml的球磨罐中,玛瑙磨球按大球占20%、中球占50%、小球占30%的质量比例装入球磨罐中,以230转/分的转速,按顺时针方向球磨50分钟,然后按逆时针方向球磨50分钟。待球磨结束后,将球磨罐里的混合物置于陶瓷方舟中,并放入石英管式炉中,接着开启真空泵抽30分钟,然后将氮气通入管式炉中,由室温经过6小时程序升温到600℃,接着经过4小时程序升温到900℃进行焙烧2小时,待冷却到室温收集得到样品,记为催化剂s3,其氮含量为25~30wt%,粒径为1~3μm。
对比例1:
将3~5g淀粉和1~2g葡萄糖溶解于60ml去离子水中,磁力搅拌30分钟,将溶液移至容积为100ml的不锈钢反应釜的内衬中,最后置于180℃的恒温烘箱中反应6小时。反应结束后,离心并用无水乙醇反复洗涤3次后,收集样品并置于80℃的真空干燥箱中干燥24小时,得到催化剂d1。
对比例2:
将3~5g葡萄糖溶解于60ml去离子水中,搅拌均匀后,将溶液移至容积为50ml的不锈钢反应釜的内衬中,最后置于190℃的电热恒温鼓风干燥箱中恒温8h。反应结束后,溶液呈混浊的黑色或红棕色,略带有焦味。反应结束后,离心并用无水乙醇反复洗涤3次后,收集样品并置于80℃的真空干燥箱中干燥24小时,得到催化剂d2。
评价例
表1为本发明实例1~3的无金属硫化氢选择性催化氧化催化剂的支构参数。从表1可知,三个样品的比表面在700~1100范围内,平均孔径在3nm左右。800℃焙烧时得到的样品比表面积最大,孔容积最大。
表1实施例1~3所述催化剂的表征的支构参数
如图1和图2所示,为本发明实例1~3所述无金属催化剂的n2物理吸脱附等温线和孔径分布图。从图中可看出,所制备的三个样品吸脱附等温线均为iv型曲线,说明这些样品中均存在介孔结构。低压区起点高,氮气吸附量大,说明样品中存在一定量的微孔。此外,三个样品的曲线均呈现h2型滞后环,说明其中均存在体心立方的介孔结构。从孔径分布图可看出,样品均存在介孔(5-8nm)和微孔(1.8nm左右)。
图3为实施例1~3和对比例1~2,所述催化剂性能评价中的h2s转化率随温度变化的曲线图。从图中可以看出本发明所制备的催化剂的h2s转化率均要比普通碳材料催化剂要高,其中最佳实施例s2催化剂在120℃时,硫化氢的转化率几乎达到100%,并随着温度的升高,转化率并没有出现明显的下降。
图4为实施例1~3和对比例1~2,所述催化剂性能评价中的催化剂对硫单质选择性随温度变化的曲线图,从图中可以看出本发明所制备的催化剂对硫单质的选择性均要明显优于普通的碳催化剂,实施例所制备的三个样品的选择性均保持在90%以上。
图5为实施例1~3和对比例1~2,所述催化剂性能评价中的硫收率随温度变化的曲线图,从图中可以看出本发明所制备催化剂的硫收率均要优于普通的碳催化剂。
图6为实施例1~3和对比例1~2,所述催化剂催化稳定性能中h2s的转化率随时间变化的曲线图,从图中可以看出本发明所制备的无金属催化剂的催化性能稳定性要明显地优于对比例催化剂的稳定性,实施例1~3所述的催化剂在反应进行25小时后,催化剂不会出现明显的失活现象,这也显示出本发明所制备催化剂优越的h2s选择性催化氧化性能。
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!