端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂的制备及应用的制作方法
本发明属于水处理技术领域,具体涉及一种端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂的制备及应用。
背景技术
随着工业的发展,水污染问题日益严重,其中阴离子污染特别是cr(vi)污染问题不容忽视。cr(vi)是环境中毒性较大且污染严重的重金属离子,由于重金属离子不可生物降解并且能长期存在于环境中。因此为了人类生活健康,有效的解决阴离子污染特别是cr(vi)污染问题是当今的重要课题。吸附法是通过材料的表面修饰、电子调控与细孔作用的不同功能实现污染物高倍数富集到吸附剂表面而实现对污染物的分离去除。目前,传统吸附剂主要存在的两大问题是:一是吸附剂表面的功能基团密度较低,吸附剂对污染物的吸附容量较小,吸附效率低;二是吸附剂的固液分离困难。
聚合物的发展依次经历了一维线性、二维交联、三维超支化等阶段。与一维线性、二维交联聚合物相比,三维超支化聚合物具有粘度低、流变性高、分子质量可控、末端官能团含量高,易接枝改性等优点。超支化聚合物富含氨基、羧基等功能基团,这不仅可以提高材料的亲水性,还可以在作为吸附剂时提高吸附剂表面功能基的密度和数量。而氧化石墨烯是一种新型的二维纳米材料,表面富含羟基、羧基及环氧基,在水体中具有一定的分散能力,但是单纯的氧化石墨烯作为吸附剂效果却不是很理想。
技术实现要素:
为解决现有技术的缺点和不足之处,本发明的首要目的在于提供一种端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂的制备方法。
本发明的另一目的在于提供一种由上述制备方法制得的端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂。
本发明的再一目的在于提供上述端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂的应用。本发明富含氨基的呈正电性点的端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂可以实现对阴离子污染物的高效去除。
本发明目的通过以下技术方案实现:
本发明通过接枝改性的方法对氧化石墨烯表面的功能基进行修饰,将超支化聚合物接枝到氧化石墨烯上可以解决吸附剂表面功能基密度低的问题,实现吸附剂对污染物的富集能力。根据已有研究可知,磁性吸附剂在外加磁场的作用下可以实现从水体中的快速分离,解决传统吸附材料固液分离困难的问题。因此,本发明在超支化聚合物改性氧化石墨烯的基础上,借助磁化试剂的作用,可以赋予吸附剂磁性,得到表面功能基密度高、易于从水体中分离的吸附剂。
一种端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂的制备方法,包括以下步骤:通过胺化试剂与丙烯酸甲酯发生迈克尔加成反应以及缩聚反应得到端氨基超支化聚合物,然后在碱性条件下将端氨基超支化聚合物接枝到磁性氧化石墨烯上,洗涤,干燥后得到所述端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂。
所述胺化试剂为二乙烯三胺、三乙烯四胺、四乙烯五胺、五乙烯六胺中的至少一种。
所述制备方法具体包括以下步骤:
(1)以甲醇或/和乙醇为溶剂,将胺化试剂与丙烯酸甲酯单体在冰浴及气体保护条件下混合均匀,得到超支化聚合物前驱体;减压蒸馏除去反应溶剂,然后经缩聚反应得到端氨基超支化聚合物;
(2)将氧化石墨烯加入到二价铁盐和三价铁盐的混合溶液中,超声分散,在搅拌及气体保护条件下将分散液加入碱性试剂溶液中,首先在70~90℃反应0.5~1h,然后冷却至40~50℃继续搅拌0.5~1h,反应结束后利用磁铁分离产物得到磁性氧化石墨烯,再将磁性氧化石墨烯加入水中经超声波分散均匀得到的磁性氧化石墨烯悬浮液;
(3)将步骤(1)得到的端氨基超支化聚合物加入碱性试剂溶液中超声分散均匀,然后加入步骤(2)得到的磁性氧化石墨烯悬浮液中,70~90℃条件下进行接枝反应12~24h,洗涤,真空冷冻干燥后得到所述端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂。
优选的,步骤(1)所述胺化试剂与丙烯酸甲酯的摩尔比为(0.5~2):1。
更优选的,步骤(1)所述胺化试剂与丙烯酸甲酯的摩尔比为1:1。
优选的,步骤(1)所述缩聚反应条件为:超支化聚合物前驱体在回流冷凝、搅拌条件下依次于120℃及140℃各反应2h。
优选的,步骤(1)、(2)中保护气体为氮气或惰性气体。
优选的,步骤(2)所述二价铁盐与三价铁盐的摩尔比为1:1~4。
优选的,步骤(2)、(3)所述的碱性试剂溶液中的碱性试剂为氢氧化钠和/或氢氧化钾。
优选的,步骤(3)所述的碱性试剂溶液中的碱性试剂与端氨基超支化聚合物的质量比为1/40~1/20。
更优选的,步骤(3)所述碱性试剂溶液中的碱性试剂与端氨基超支化聚合物的质量比为3/80。
优选的,步骤(3)所述的磁性氧化石墨烯悬浮液中的氧化石墨烯与端氨基超支化聚合物的质量比为1/20~1/5。
更优选的,步骤(3)所述的磁性氧化石墨烯悬浮液中的氧化石墨烯与端氨基超支化聚合物的质量比为1/10。
本发明还提供了一种由上述制备方法制得的端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂。
本发明所述的端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂在去除废水中阴离子污染物的应用,所述废水包括有印染废水、含铬废水或焦化废水。
优选的,所述阴离子污染物为cr(vi)、no3-或so42-。
与现有技术相比,本发明具有以下优点及有益效果:
(1)本发明所制备的端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂含有高密度的氨基功能基,而显正电性氨基功能基可以实现对显负电性的阴离子污染物的高度富集及快速去除,因此吸附剂对阴离子污染物具有较高的吸附容量。
(2)本发明将经“一步法”缩聚反应得到富含高密度氨基的端氨基超支化聚合物引入比表面积大且富含羟基、羧基、环氧基的氧化石墨烯,充分结合了超支化大分子与氧化石墨烯的优势。本发明所制备的端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂是固体吸附剂,结构稳定,不易分解,可以重复利用,降低了生产成本。
(3)本发明制备的端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂用于废水中阴离子的吸附去除时,在外加磁场的作用下,吸附剂可以非常容易地从溶液中分离,回收方便。经碱性试剂洗脱后,阴离子污染物可脱附下来,实现吸附剂的再生回用。
附图说明
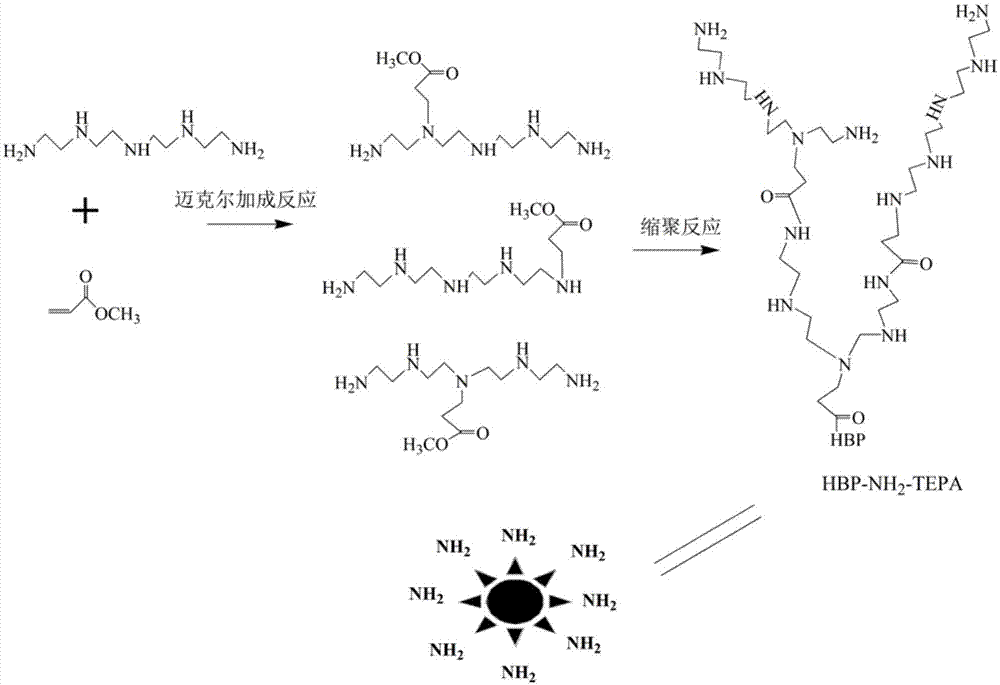
图1为实施例3步骤(1)制备端氨基超支化聚合物的反应式。
图2为实施例3步骤(6)制备石墨烯接枝端氨基超支化聚合物磁性阴离子吸附剂的反应式。
图3为实施例3制备的端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂的可能结构式。
图4为氧化石墨烯、实施例1-3制备的端氨基超支化聚合物接枝氧化石墨烯的zeta电位随ph值的变化图。
图5为实施例1-3中制备的端氨基超支化聚合物接枝氧化石墨烯的元素组成分析图(通过xps测试得到)。
图6为实施例4中制备的端氨基超支化聚合物接枝氧化石墨烯阴离子吸附剂吸附cr(vi)的元素组成图(通过xps测试得到)。
图7为实施例4中制备的端氨基超支化聚合物接枝氧化石墨烯的再生回用性能图。
其中,图4-5中,go-hbp-nh2-deta为实施例1制备的端氨基超支化聚合物接枝氧化石墨烯(以二乙烯三胺为单体),go-hbp-nh2-teta为实施例2制备的端氨基超支化聚合物接枝氧化石墨烯(以三乙烯四胺为单体),go-hbp-nh2-tepa为实施例3制备的端氨基超支化聚合物接枝氧化石墨烯(以四乙烯五胺为单体)。
具体实施方式
下面结合实施例和附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
实施例1
1.一种端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂的制备方法,包括步骤如下:
(1)将0.5mol二乙烯三胺与50ml无水甲醇混合,置于0℃下,通入n2,搅拌10min后,将1mol丙烯酸甲酯(ma)与50ml无水甲醇的混合溶液缓慢滴加至二乙烯三胺与甲醇的混合溶液中,二乙烯三胺与丙烯酸甲酯(ma)发生迈克尔加成反应,得到淡黄色液体即为超支化聚合物前驱体(中间产物)。然后将所得前驱体转移到旋转蒸发仪的圆底烧瓶中,在循环水式真空泵抽真空条件下,在60℃保持2h,减压蒸馏去除溶剂甲醇;再将温度依次升至120℃、140℃保持搅拌各减压缩聚反应2h,制得hbp-nh2超支化聚合物。
(2)将20ml浓度0.5mol/l的naoh溶液加入三口烧瓶中,并在80℃不断搅拌。将0.001molfecl3·6h2o及0.0005molfeso4·7h2o加入到30ml乙醇溶液中(1/1,v/v),然后加入3g氧化石墨烯,室温下超声分散30min;将超声分散后的混合液逐滴加入到上述氢氧化钠溶液中,氮气保护下不断搅拌,与此同时,可见黑色沉淀。80℃反应30min后冷却至50℃,然后继续搅拌1h。
(3)将步骤(2)反应完成后所得黑色沉淀利用永久磁铁分离黑色沉淀,然后分别用无水乙醇、蒸馏水洗涤以去除残余杂质,在50℃条件下真空干燥24h,得到磁性氧化石墨烯。
(4)将0.4g步骤(3)所得磁性氧化石墨烯分散至100ml蒸馏水中,超声分散30min。
(5)将0.15g的koh溶于50ml蒸馏水中,然后加入4g步骤(1)所得hbp-nh2超支化聚合物,超声分散30min。
(6)将步骤(4)和步骤(5)所得液体样品混合,然后在80℃条件下搅拌12h。将利用永久磁铁将产物分离,然后用无水乙醇、蒸馏水洗涤数次,以去除未反应的hbp-nh2,真空冷冻干燥48h,得到端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂(go-hbp-nh2)。
2.将制备得到的端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂用于cr(vi)的吸附,吸附条件:cr(vi)溶液初始浓度50mg/l,吸附剂用量10mg,cr(vi)溶液体积50ml,温度25℃,ph=2,吸附时间6h。吸附完成后,用0.45μm滤膜过滤上层清液,用原子吸收对残余cr(vi)浓度进行检测,根据相关计算得到吸附剂对cr(vi)的吸附容量为245.1mg/g。
3.将制备得到的端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂用于no3-及so42-的吸附,经离子色谱测定,吸附剂对no3-及so42-的吸附容量分别可达16.7mg/g、23.5mg/g。
实施例2
1.一种端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂的制备方法,包括步骤如下:
(1)将0.5mol三乙烯四胺与50ml无水甲醇混合,置于0℃下,通入n2,搅拌10min后,将1mol丙烯酸甲酯(ma)与50ml无水甲醇的混合溶液缓慢滴加至三乙烯四胺与甲醇的混合溶液中,三乙烯四胺与丙烯酸甲酯(ma)发生迈克尔加成反应,得到淡黄色液体即为超支化聚合物前驱体(中间产物)。然后将所得前驱体转移到旋转蒸发仪的圆底烧瓶中,在循环水式真空泵抽真空条件下,在60℃保持2h,减压蒸馏去除溶剂甲醇;再将温度依次升至120℃、140℃保持搅拌各减压缩聚反应2h,制得hbp-nh2超支化聚合物。
(2)将20ml的0.5mol/lnaoh溶液加入三口烧瓶中,并在70℃不断搅拌。将0.002molfecl3·6h2o及0.0005molfeso4·7h2o加入到30ml乙醇溶液中(1/1,v/v),然后加入3g氧化石墨烯,室温下超声分散30min;将超声分散后的混合液逐滴加入到上述氢氧化钠溶液中,氮气保护下不断搅拌,与此同时,可见黑色沉淀。70℃反应1h后冷却至40℃,然后继续搅拌0.5h。
(3)将步骤(2)反应完成后所得黑色沉淀利用永久磁铁分离黑色沉淀,然后分别用无水乙醇、蒸馏水洗涤以去除残余杂质,在50℃条件下真空干燥24h,得到磁性氧化石墨烯。
(4)将0.3g步骤(3)所得磁性氧化石墨烯分散至100ml蒸馏水中,超声分散30min。
(5)将0.15g的naoh溶于50ml蒸馏水中,然后加入4g步骤(1)所得hbp-nh2超支化聚合物,超声分散30min,
(6)将步骤(4)和步骤(5)所得液体样品混合,然后在70℃条件下搅拌24h。将利用永久磁铁将产物分离,然后用无水乙醇、蒸馏水洗涤数次,以去除未反应的hbp-nh2,真空冷冻干燥48h,得到端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂(go-hbp-nh2)。
2.将制备得到的端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂用于cr(vi)的吸附,吸附条件:cr(vi)溶液初始浓度50mg/l,吸附剂用量10mg,cr(vi)溶液体积50ml,温度25℃,ph=2,吸附时间6h。吸附完成后,用0.45μm滤膜过滤上层清液,用原子吸收对残余cr(vi)浓度进行检测,根据相关计算得到吸附剂对cr(vi)的吸附容量为257.3mg/g。
3.将制备得到的端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂用于no3-及so42-的吸附,经离子色谱测定,吸附剂对no3-及so42-的吸附容量分别可达25.1mg/g、31.4mg/g。
实施例3
1.一种端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂的制备方法,包括步骤如下:
(1)将1mol四乙烯五胺与50ml无水甲醇混合,置于0℃下,通入n2,搅拌10min后,将0.5mol丙烯酸甲酯(ma)与50ml无水甲醇的混合溶液缓慢滴加至四乙烯五胺与甲醇的混合溶液中,四乙烯五胺与丙烯酸甲酯(ma)发生迈克尔加成反应,得到淡黄色液体即为超支化聚合物前驱体(中间产物)。然后将所得前驱体转移到旋转蒸发仪的圆底烧瓶中,在循环水式真空泵抽真空条件下,在60℃保持2h,减压蒸馏去除溶剂甲醇;再将温度依次升至120℃、140℃保持搅拌各减压缩聚反应2h,制得hbp-nh2超支化聚合物。
(2)将20ml的0.5mol/lnaoh溶液加入三口烧瓶中,并在90℃不断搅拌。将0.0005molfecl3·6h2o及0.0005molfeso4·7h2o加入到30ml乙醇溶液中(1/1,v/v),然后加入3g氧化石墨烯,室温下超声分散30min;将超声分散后的混合液逐滴加入到上述氢氧化钠溶液中,氮气保护下不断搅拌,与此同时,可见黑色沉淀。90℃反应30min后冷却至50℃,然后继续搅拌45min。
(3)将步骤(2)反应完成后所得黑色沉淀利用永久磁铁分离黑色沉淀,然后分别用无水乙醇、蒸馏水洗涤以去除残余杂质,在50℃条件下真空干燥24h,得到磁性氧化石墨烯。
(4)将0.2g步骤(3)所得磁性氧化石墨烯分散至100ml蒸馏水中,超声分散30min。
(5)将0.15g的naoh溶于50ml蒸馏水中,然后加入4g步骤(1)所得hbp-nh2超支化聚合物,超声分散30min。
(6)将步骤(4)和步骤(5)所得液体样品混合,然后在90℃条件下搅拌18h。将利用永久磁铁将产物分离,然后用无水乙醇、蒸馏水洗涤数次,以去除未反应的hbp-nh2,真空冷冻干燥48h,得到端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂(go-hbp-nh2)。
2.将制备得到的端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂用于cr(vi)的吸附,吸附条件:cr(vi)溶液初始浓度50mg/l,吸附剂用量10mg,cr(vi)溶液体积50ml,温度25℃,ph=2,吸附时间6h。吸附完成后,用0.45μm滤膜过滤上层清液,用原子吸收对残余cr(vi)浓度进行检测,根据相关计算得到吸附剂对cr(vi)的吸附容量为300.8mg/g。
3.将制备得到的端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂用于no3-及so42-的吸附,经离子色谱测定,吸附剂对no3-及so42-的吸附容量分别可达32.5mg/g,40.1mg/g。
实施例4
1.一种端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂的制备方法,包括步骤如下:
(1)将0.5mol五乙烯六胺与50ml无水乙醇混合,置于0℃下,通入n2,搅拌10min后,将0.5mol丙烯酸甲酯(ma)与50ml无水甲醇的混合溶液缓慢滴加至五乙烯六胺与甲醇的混合溶液中,五乙烯六胺与丙烯酸甲酯(ma)发生迈克尔加成反应,得到淡黄色液体即为超支化聚合物前驱体(中间产物)。然后将所得前驱体转移到旋转蒸发仪的圆底烧瓶中,在循环水式真空泵抽真空条件下,在60℃保持2h,减压蒸馏去除溶剂乙醇;再将温度依次升至120℃、140℃保持搅拌各减压缩聚反应2h,制得hbp-nh2超支化聚合物。
(2)将20ml的0.5mol/lnaoh溶液加入三口烧瓶中,并在80℃不断搅拌。将0.001molfecl3·6h2o及0.0005molfeso4·7h2o加入到30ml乙醇溶液中(1/1,v/v),然后加入3g氧化石墨烯,室温下超声分散30min;将超声分散后的混合液逐滴加入到上述氢氧化钠溶液中,氮气保护下不断搅拌,与此同时,可见黑色沉淀。80℃反应45min后冷却至50℃,然后继续搅拌1h。
(3)将步骤(2)反应完成后所得黑色沉淀利用永久磁铁分离黑色沉淀,然后分别用无水乙醇、蒸馏水洗涤以去除残余杂质,在50℃条件下真空干燥24h,得到磁性氧化石墨烯。
(4)将0.4g步骤(3)所得磁性氧化石墨烯分散至100ml蒸馏水中,超声分散30min。
(5)将0.15g的koh溶于50ml蒸馏水中,然后加入2g步骤(1)所得hbp-nh2超支化聚合物,超声分散30min。
(6)将步骤(4)和步骤(5)所得液体样品混合,然后在80℃条件下搅拌12h。将利用永久磁铁将产物分离,然后用无水乙醇、蒸馏水洗涤数次,以去除未反应的hbp-nh2,真空冷冻干燥48h,得到端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂(go-hbp-nh2)。
2.将制备得到的端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂用于cr(vi)的吸附,吸附条件:cr(vi)溶液初始浓度50mg/l,吸附剂用量10mg,cr(vi)溶液体积50ml,温度25℃,ph=2,吸附时间6h。吸附完成后,用0.45μm滤膜过滤上层清液,用原子吸收对残余cr(vi)浓度进行检测,根据相关计算得到吸附剂对cr(vi)的吸附容量为300.8mg/g。
3.将制备得到的端氨基超支化聚合物接枝氧化石墨烯磁性阴离子吸附剂用于no3-及so42-的吸附,经离子色谱测定,吸附剂对no3-及so42-的吸附容量分别可达31.4mg/g,42.8mg/g。
根据本发明制备合成端氨基超支化聚合物接枝磁性氧化石墨烯阴离子吸附剂的设计思路(以实施例3为例,如图1、图2和图3所示),所得吸附剂含有大量带正电荷的氨基功能基团。通过不同ph值条件下zeta测试(图4),可得go-hbp-nh2-deta、go-hbp-nh2-teta及go-hbp-nh2-tepa三种吸附剂的零电位点分别为6.0、6.5、及6.7,这表明所得三种吸附剂含有大量的具有正电性的功能基团。当溶液ph值小于吸附剂的零电位点时,吸附剂显正电性,可以吸附溶液中的阴离子污染物;当溶液ph值大于吸附剂的零电位点时,吸附剂显负电性,可以吸附溶液中的阳离子污染物。以go-hbp-nh2-tepa为例,当溶液ph值为2时,go-hbp-nh2-tepa表面带正电荷,可以实现去cr(vi)的高效吸附。
胺化试剂的选择对所得端氨基超支化聚合物接枝氧化石墨烯阴离子吸附剂中氮元素的含量具有十分重要的影响,进而影响氨基功能基的数量和密度。如图5所示,当胺化试剂分别为二乙烯三胺、三乙烯四胺及四乙烯五胺时,所得阴离子吸附剂中氮元素的含量分别为7.21wt%、10.20wt%及12.43%。
将端氨基超支化聚合物接枝氧化石墨烯阴离子吸附剂用于cr(vi)的吸附,吸附反应完成后,吸附后的吸附剂将含有一定量的铬元素。如图6所示,通过xps测试得到的元素含量分析发现,吸附剂中铬元素含量为7.79%,这表明吸附反应成功进行。
吸附剂的再生回用性能将在一定程度上决定吸附剂的价格,是决定吸附剂可否用于实际应用的关键要素之一。吸附实验完成后,采用0.05mol/l的naoh溶液对吸附剂进行洗脱再生。回用实验发现,经5次回用后,所得端氨基超支化聚合物接枝氧化石墨烯阴离子吸附剂(以五乙烯六胺为胺化试剂)的再生率仍可达80%以上,如图7所示,这表明本发明所得吸附剂具有良好的再生回用特性。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!