一种手性多酸基金属有机框架材料及其制备方法和应用与流程
本发明属于多金属氧酸盐功能材料制备技术领域,具体涉及一种手性多酸基金属有机框架材料及其制备方法和应用。
背景技术:
环氧化合物是有机合成的重要中间体和化工原料,“十一五”是我国环氧丙烷行业蓬勃发展,成为环氧丙烷全球最大产销国,2010年产能达到154.5万吨,产量143.76万吨。随着聚醚多元醇行业、高品质电子产品、航空航天产品制造的大量需求,我国环氧丙烷产品需求仍在快速发展,预测到2020年消费量很快会达到275.6万吨。尤其是具有光学活性的环氧化物是手性药物合成的重要中间体,通过环氧化物的选择性开环和官能团转换等反应,可以合成出许多有价值的医药、农药。将来源于石油化石资源的碳氢化合物等化工原料经选择性氧化转化为相应的具有高附加值的手性含氧化合物,使其作为制备聚合物单体以及农药和医药品的基本原料,是现代石油化工行业的主要反应过程之一。sharpless环氧化(ae)反应是手性技术的重大突破,但该反应具有一定的局限性如仅适用于烯丙醇底物而且使用的氧化剂tbhp不太稳定。随后,不同的手性催化剂被开发对sharpless体系进行了补充和完善,如jacobsen催化剂以naocl为氧化剂用于非官能团的烯烃环氧化(j.am.chem.soc.,1990,112,2801)、groves手性卟啉络合催化剂以phio为氧化剂用于苯乙烯类底物(j.am.chem.soc.,1991,113,6865)、杨丹和史一安的手性酮催化剂分别以khso5和hclo4为氧化剂用于取代烯烃(j.am.chem.soc.,1996,118,491;1996,118,11311;j.am.chem.soc.,1997,119,11224)、julia-colonna催化剂以脲素过氧化氢络合物为氧化剂用于-不饱和酮(醛)(chem.commun.,1997,739)。虽然以上均相手性催化具有高效、高对映选择性和反应条件温和等特点,但是要实现这些催化反应在工业上的应用,必须首先解决昂贵催化剂的回收利用这个严重的问题,而且这些催化体系主要是基于热催化反应过程,需要有机过酸、有机过氧化物等作氧化剂,存在过程成本高、腐蚀性、危险性、毒性、严重的环境污染和苛刻反应条件等弊端。
技术实现要素:
本发明所要解决的技术问题在于针对上述现有技术的不足,提供一种手性多酸基金属有机框架材料,该材料属于多功能无机多孔材料,其具有不对称光催化氧化技术,该技术是一种通过可见光驱动光敏催化剂至激发态后与底物发生氧化还原从而完成相关自由基类型的反应,而本发明的手性多酸基金属有机框架材料能为不对称光催化氧化反应提供更好的技术。
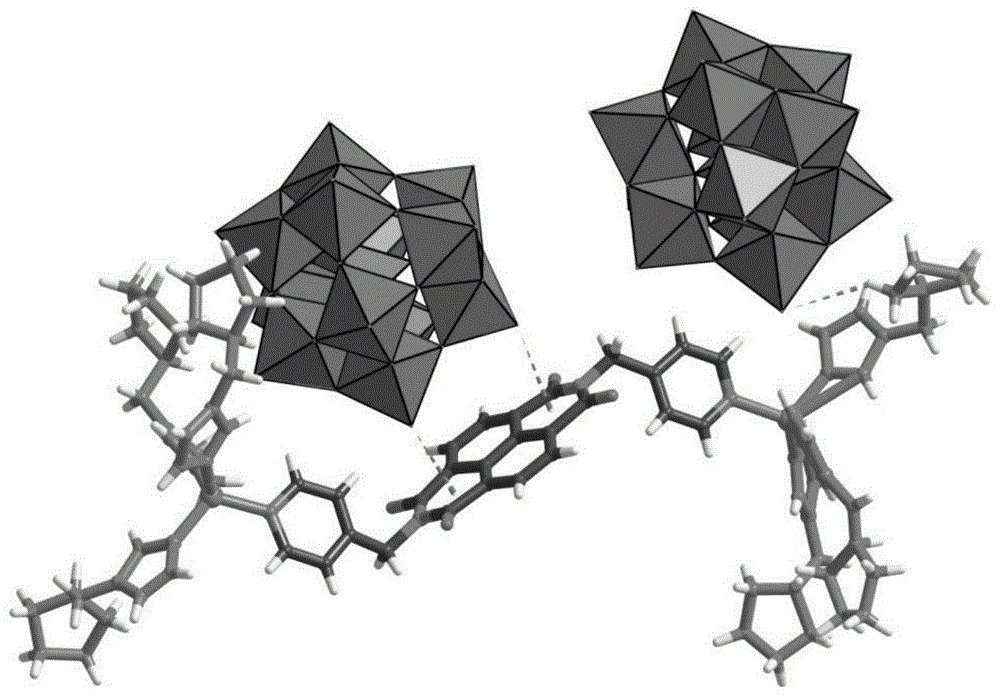
为解决上述技术问题,本发明采用的技术方案是:一对具有光催化功能的手性多酸基金属有机框架材料,其特征在于,所述多酸基金属有机框架材料的化学式为:c74h101b2zn2n22o84w24,记为znw-dpndi-pyis,分别记为znw-dpndi-pyi1和znw-dpndi-pyi2,其中dpndi为n,n’-二(4-甲基吡啶)萘酰亚胺,作为桥连配体和光敏剂;pyi为l-吡咯烷-2-咪唑,作为手性配体和电子供体;所述多酸基金属有机框架材料的多酸阴离子(poms)作为氧化催化功能基团;所属晶体均属于单斜晶系,所述znw-dpndi-pyi1和znw-dpndi-pyi2的空间群均为p2(1);znw-dpndi-pyi1的单晶胞结构图如图1所示。
本发明还提供了多酸基金属有机框架材料的制备方法,所述znw-dpndi-pyi1的制备方法为:分别称取45mg、0.15mmol的zn(no3)2·6h2o,157.8mg、0.05mmol的k5[bw12o40]·5h2o,23.4mg、0.15mmol的dpndi和24.5mg、0.1mmol的l-bcip(l-n-叔丁氧羰基-2-咪唑-1-吡咯烷)于小玻璃瓶中,加入4.0ml蒸馏水、2.0ml甲醇溶解,搅拌过夜,得到悬浊溶液,将所述悬浊溶液转移至25ml的高压反应釜中,放入烘箱,加热到120℃反应五天,冷却至室温后取出反应产物,用蒸馏水反复洗涤,得到白黄色棒状晶体znw-dpndi-pyi1,所述znw-dpndi-pyi1的产率为60%;
所述znw-dpndi-pyi2的制备方法为:分别称取45mg、0.15mmol的zn(no3)2·6h2o,157.8mg、0.05mmol的k5[bw12o40]·5h2o,23.4mg、0.15mmol的dpndi和24.5mg、0.1mmol的d-bcip于小玻璃瓶中,加入4.0ml蒸馏水、2.0ml甲醇溶解,搅拌过夜,得到悬浊溶液,将所述悬浊溶液转移至25ml的高压反应釜中,放入烘箱,加热到120℃反应四天,冷却至室温后取出反映产物,用蒸馏水反复洗涤,得到白黄色棒状晶体znw-dpndi-pyi2,所述znw-dpndi-pyi2的产率为60%。
原料l-bcip为l-n-叔丁氧羰基-2-咪唑-1-吡咯烷,或d-bcip为d-n-叔丁氧羰基-2-咪唑-1-吡咯烷,其参照文献(s.luo,mi,x.l.zhang,s.liu,h.xu,j.p.cheng,angew.chem.int.ed.2006,45,3093–3097)中公开的方法合成得到。
原料dpndi为n,n’-二(4-甲基吡啶)萘酰亚胺,其参照文献(g.b.li,j.m.liu,z.q.yu,w.wang,c.y.su,inorg.chem.2009,48,8659–8661)中公开的方法合成得到。
原料k5bw12o40为keggin型多酸盐k5bw12o40,其参照文献(c.rocchiccioli-deltcheff,m.fournier,r.franck,r.thouvenot,inorg.chem.1983,22,207–216)中公开的方法合成得到。
本发明还提供了一种手性多酸基金属有机框架材料应用于胺的c-n耦合和烯烃环氧化的光催化反应,具体应用为:以znw-dpndi-pyis为催化剂,在可见光照射条件下,以o2为单一氧化源,应用于胺的c-n耦合和烯烃环氧化的光催化反应中。
本发明的原理:dpndi是萘酰亚胺的衍生物,是一类具有光化学,光物理,电化学及催化特能力的多功能π-缺电子有机配体,由于其二酰亚胺上的n原子的可调控性及萘环中电子转移的可还原性,使得萘酰亚胺成为了一类极佳的多功能金属-有机框架材料(metal-organicframeworks,mofs)的构筑块。多金属氧酸盐(多酸,polyoxometalates,poms)具有高质子酸性、低温高活性、好的热稳定性及杂多酸独特的“假液相”反应场等特点。其中,多酸具有丰富的氧化还原位点及光催化活性,有光诱导电子转移的潜能,进而产生光致变色现象。脯氨酸是一种最为常见的手性配体,通过氢键作用可实现高效的不对称催化,这是近年有机小分子催化的另一亮点,同时其氮原子的富电性使得pyi也可作为一种供电子体。我们将具有良好催化烯烃环氧化功能的[bw12o40]5-多酸阴离子、可充当光敏剂的dpndi及具有立体定向性的手性配体pyi同时引入到精心设计mofs中,得到一对对映体催化剂znw-dpndi-pyi1和znw-dpndi-pyi2。dpndi吸收可见光后,可以通过连续光诱导电子转移过程产生dpndi·-和dpndi·-*,然后电子转移给[bw12(vi)o40]5-形成具有还原性的杂多兰[bw11w(v)o40]6-,pyi提供电子给激发态的dpndi·-,同时手性pyis将作为协同催化位点,形成驱动多相不对称反应的关键反应中心。将poms、手性配体同时引入到mof结构中,构筑具有多重催化位点的手性多酸基金属有机框架材料mofs,既能发挥各组分的优点,又可实现催化位点的协同催化。
本发明与现有技术相比具有以下优点:
1、本发明提供的znw-dpndi-pyis为多酸基金属有机框架材料,属于多功能无机多孔材料,其具有不对称光催化氧化技术,该技术是一种通过可见光驱动光敏催化剂至激发态后与底物发生氧化还原从而完成相关自由基类型的反应。多功能无机多孔材料znw-dpndi-pyis能为不对称光催化氧化反应提供更好的技术。
2、本发明的多酸基金属有机框架材料可通过x-射线单晶衍射准确地了解其结构特征。
3、本发明以发展功能为导向的手性催化剂的可控组装和定向制备为核心目标,合理利用配位导向作用,构建立体结构稳定、性能良好的多功能手性金属有机框架结构反应平台。通过发挥金属有机框架结构内部孔道的特殊空间限制,利用多重催化位点之间的协同作用,实现从简单烃类原料高效构建结构复杂的手性环氧化合物的过程。而且,我国钨、钼的储量居世界首位,为以后的实际应用提供了发展空间。
4、本发明的制备方法和应用具有高效性、原子经济性和绿色化等特点,光催化合成的策略,研究催化体系在动力学上的控制,从而选择出具有最优反应时间、转化率与最大收率的反应条件;通过调控“催化位点–底物”之间立体、电子效应匹配,实现手性催化的立体选择性控制。
下面结合附图和实施例对本发明作进一步详细说明。
附图说明
图1为本发明实施例1制备的znw-dpndi-pyi1单胞结构图。
图2是本发明znw-dpndi-pyi1和znw-dpndi-pyi2的合成及不对称催化位点示意图。
图3是本发明实施例1制备的znw-dpndi-pyi1(a)和znw-dpndi-pyi2(b)的cd光谱图。
图4为本发明实施例1制备的znw-dpndi-pyi1沿c轴方向的三维堆积图。
图5为本发明实施例1制备的znw-dpndi-pyi1的固体紫外可吸收光谱图、荧光图及xps谱图,(a)为znw-dpndi-pyi1的固体紫外;(b)和(d)为znw-dpndi-pyi1的xps谱图;(c)为znw-dpndi-pyi1的荧光图。
图6为芳基烯烃环氧化反应表达式。
图7为催化烯烃环氧化反应表达式。
图8为催化机理推测图。
具体实施方式
实施例1
本实施例手性多酸基金属有机框架材料的化学式为:c74h101b2zn2n22o84w24,记为znw-dpndi-pyis,根据材料手性又分别记为znw-dpndi-pyi1和znw-dpndi-pyi2。
本实施例znw-dpndi-pyi1的制备方法为:分别称取45mg、0.15mmol的zn(no3)2·6h2o,157.8mg、0.05mmol的k5[bw12o40]·5h2o,23.4mg、0.15mmol的dpndi和24.5mg、0.1mmol的l-bcip于小玻璃瓶中,加入4.0ml蒸馏水、2.0ml甲醇溶解,搅拌过夜,得到悬浊溶液,将所述悬浊溶液转移至25ml的高压反应釜中,放入烘箱,加热到120℃反应五天,冷却至室温后取出反应产物,用蒸馏水反复洗涤,得到白黄色棒状晶体znw-dpndi-pyi1,所述znw-dpndi-pyi1的产率为60%;
本实施例znw-dpndi-pyi2的制备方法为:分别称取45mg、0.15mmol的zn(no3)2·6h2o,157.8mg、0.05mmol的k5[bw12o40]·5h2o,23.4mg、0.15mmol的dpndi和24.5mg、0.1mmol的d-bcip于小玻璃瓶中,加入4.0ml蒸馏水、2.0ml甲醇溶解,搅拌过夜,得到悬浊溶液,将所述悬浊溶液转移至25ml的高压反应釜中,放入烘箱,加热到120℃反应四天,冷却至室温后取出反映产物,用蒸馏水反复洗涤,得到白黄色棒状晶体znw-dpndi-pyi2,所述znw-dpndi-pyi2的产率为60%。
本实施例中,原料l-bcip为l-n-叔丁氧羰基-2-咪唑-1-吡咯烷,或d-bcip为d-n-叔丁氧羰基-2-咪唑-1-吡咯烷,其参照文献(s.luo,mi,x.l.zhang,s.liu,h.xu,j.p.cheng,angew.chem.int.ed.2006,45,3093–3097)中公开的方法合成得到。
本实施例中,原料dpndi为n,n’-二(4-甲基吡啶)萘酰亚胺,其参照文献(g.b.li,j.m.liu,z.q.yu,w.wang,c.y.su,inorg.chem.2009,48,8659–8661)中公开的方法合成得到。
本实施例中,原料k5bw12o40为keggin型多酸盐k5bw12o40,其参照文献(c.rocchiccioli-deltcheff,m.fournier,r.franck,r.thouvenot,inorg.chem.1983,22,207–216)中公开的方法合成得到。
本实施例中,原料zn(no3)2选自市售的分析纯zn(no3)2。
对制备的多酸基金属有机框架材料的结构进行如下分析:
图3是本实施例1制备的znw-dpndi-pyi1(a)和znw-dpndi-pyi2(b)的cd光谱图,对图3分析结果表明znw-dpndi-pyi1和znw-dpndi-pyi2是一对对映体结构。以znw-dpndi-pyi1为例详细介绍晶体结构。晶体学相互独立的两个的锌原子展现相同的配位环境,均为畸变的四面体构型,由1个桥联配体dpndi连接起来,并分别与3个来自于手性配体l-pyi的原子配,两个[bw12o40]5–均处于游离状态(如图1所示)。其中一个[bw12o40]5-恰好落于dpndi萘环的正上方,中心距离为
对本发明的多酸基金属有机框架材料的应用通过实施例2~实施例4进行详细阐述。
实施例2
本实施例以苯乙烯为底物,o2为氧化剂,加入0.1%mol的znw-dpndi-pyi1或znw-dpndi-pyi2,在可见光照射下反应12h,实现了非均相不对称催化烯烃环氧化反应,反应方程式如图6所示,反应条件和反应结果如表1所示,苯乙烯的选择性氧化产率达92%,转化为产物氧化苯乙烯,同时ee值高达45%,底物可以扩展到苯乙烯衍生物,如4-氯基苯乙烯、2-氯苯乙烯、4-甲基苯乙烯。
实施例3
如表1中选择多种底物进行催化氧化反应,其中选用3,5-二叔丁基-4-乙烯基联苯(即表1中的叔丁基苯乙烯)作为底物时,在与实施例2相同条件下,不对称催化环氧化反应产率不足10%。我们推测这是由于该底物体积过大,无法进入znw-dpndi-pyi1的孔道中进行反应,仅有少量底物吸附在znw-dpndi-pyi1表面反应。这一现象不仅体现了pomofs择形催化的优势,也证明了本实验中的不对称催化环氧化反应确实是发生在pomofs孔道之中,高效高选择性是pomofs孔道中多个催化位点协同作用的结果。
表1芳基烯烃环氧化反应的转化率和选择性
表1中反应方程式可通过图6表示,反应条件:底物烯烃5mmol,znw-dpndi-pyi1(10mg,12.5μmol,0.02mol%),ch3cn3ml;空气气氛下,10w白色led灯,室温,反应时间12h。转化率和对环氧化物的选择性由粗产物的核磁共振氢谱测定。
实施例4
本实施例选用不同氧化剂作为对照实验,n2条件下不反应。通过引入一系列捕获剂,考察了氧气主要被活化为单线态氧(1o2)和超氧自由基(o2-),进一步形成羟基自由基(oh)。对该反应机理进行推测,如图7所示。结果表明在该结构中,各个催化功能基团通过协同作用高效率和高选择性的催化烯烃不对称环氧化过程。
表2不同氧化剂、催化剂和自由基清除剂光催化氧化苯乙烯的转化率和选择性
“-”表示未测出该项产物。
表2中反应方程式如图7所示,反应条件:底物:苯乙烯,5mmol,ch3cn,3ml;10w白色led灯,室温,12h,(条目1-6:znw-dpndi-pyi1,12.5μmol;空气,1个大气压o2或n2;条目4-6:空气和5mgdabco,5mgbq或0.5mlipa。条目7-8,dpnd,10mg;k5[bw12o40],10mg,/k5[bw12o40]/dpndi/pyi,10mg。转化率和对环氧化物的选择性由粗产物的核磁共振氢谱测定。
以znw-dpndi-pyis为催化剂,在可见光照射条件下,以o2为单一氧化源,应用于胺的c-n耦合和烯烃环氧化的光催化反应中,催化机理推测如图8所示,dpndi是一类具有光化学,光物理,电化学及催化特能力的多功能π-缺电子有机配体,由于其二酰亚胺上的n原子的可调控性及萘环中电子转移的可还原性,使得萘酰亚胺成为了一类极佳的多功能金属-有机框架材料的构筑块。pyi的氮原子的富电性使其作为一种供电子体,[bw12o40]5-多酸阴离子具有良好催化烯烃环氧化功能、dpndi可充当光敏剂,pyi具有立体定向性的手性配体,dpndi吸收可见光后,可以通过连续光诱导电子转移过程产生dpndi·-和dpndi·-*,然后电子转移给[bw12(vi)o40]5-形成具有还原性的杂多兰[bw11w(v)o40]6-,pyi提供电子给激发态的dpndi·-,同时手性pyis将作为协同催化位点,形成驱动多相不对称反应的关键反应中心。
以上所述,仅是本发明的较佳实施例,并非对本发明作任何限制。凡是根据发明技术实质对以上实施例所作的任何简单修改、变更以及等效变化,均仍属于本发明技术方案的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!