一种含硫氧化物和NO的气体的处理方法与流程
本发明涉及一种含no气体的处理方法,更进一步说涉及含硫氧化物、氧气和no的气体中no的处理方法。
背景技术:
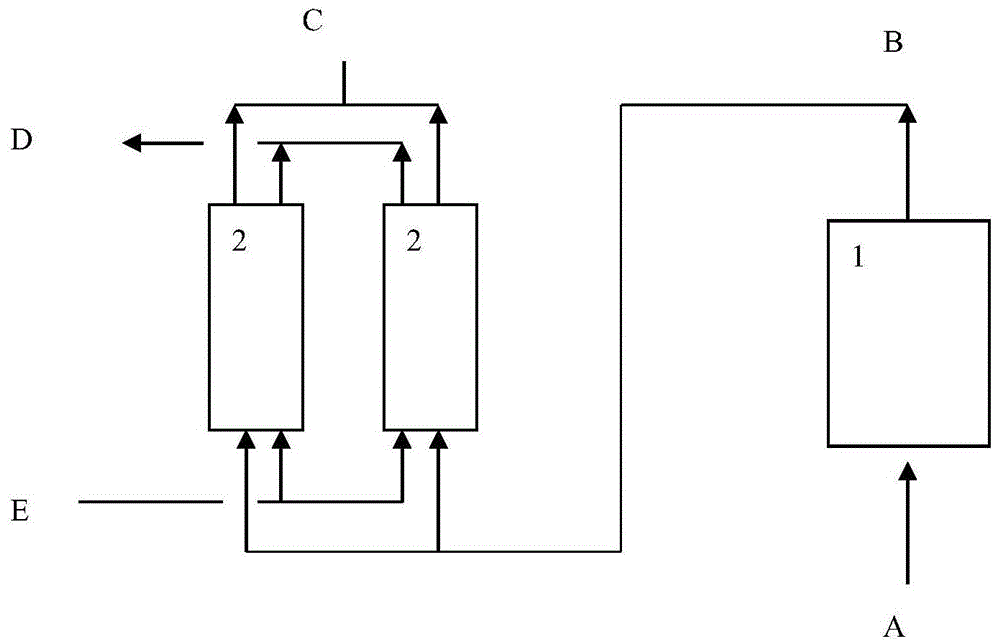
:煤炭、石油等在燃烧过程中产生的硫氧化物(sox:so3和so2)或氮氧化物(nox)是造成酸雨的主要根源,是雾霾的前驱体。工业生产过程中,尤其是在一些含有热处理步骤(干燥、焙烧、燃烧等)的生产过程中,也会产生含有污染物的尾气或烟气等废气。例如,在催化裂化(fcc)过程中,原料中的部分硫、氮化合物在提升管反应过程中进入焦炭沉积于待生催化剂上,待生剂进入再生器烧焦再生时,这些焦炭中的硫、氮化合物氧化生成sox、nox等烟气污染物。再如,催化剂生产等无机化工过程中,原材料中含有的硫酸盐、硝酸盐等加热分解时会产生含有sox、nox污染物和有害气体的尾气。这些含污染物和有害气体的烟气和尾气需经过净化处理后才能达标排放。sox可以利用酸碱反应来脱除,反应简单,操作窗口宽;no既不具备水溶性,也没有酸碱性,很难脱除。而烟气中,nox中的no占比通常在95%左右,因此nox的脱出关键在于no的脱除。现有的no脱除方法主要包括三种:(1)选择性催化还原(scr)法,即在催化剂的作用下,在含有no的气体中喷入还原剂氨或尿素,把其中的nox还原成n2和h2o;(2)no直接高温分解,这一过程在催化剂存在下进行;(3)no催化氧化法,将no转化为能被碱性溶液吸收的no2,进行脱除。其中,scr方法催化剂成本高,并且存在氨逃逸等二次污染等问题,且存在副产物硫酸铵堵塞烟道的情况;no高温分解虽然不存在二次污染等情况,但是催化剂活性易被抑制,尤其受烟气中氧含量影响严重等问题;而no催化氧化能够在适宜的反应温度下借助烟气中的氧气将no氧化为no2后将no2脱除。技术实现要素:本发明的发明人研究发现,现有no氧化方法,no氧化催化剂在使用一段时间后存在no转化率下降的问题,本发明的发明人研究认为,这是由于硫氧化物的存在,使no氧化催化剂中毒导致。为了解决上述问题,本发明提出以下技术方案:技术方案1.一种含硫氧化物、氧气和no的气体中no的氧化方法,包括,将含硫氧化物、氧气和no的气体与no氧化催化剂接触反应,其中,所述的no氧化催化剂包括镁组分和氧化活性组分。技术方案2.根据技术方案1所述的方法,其中,所述的no氧化催化剂包括镁组分、氧化活性组分和基质;以所述催化剂的干基重量为基准,所述的活性组分的含量为0.4~40重量%例如5~40重量%,镁组分的含量为0.3-28.5重量%例如2~25重量%,基质含量为42~90.725重量%例如45~85重量%。技术方案3.根据技术方案1或2所述的方法,其中,所述的基质组分包括介孔氧化硅材料和/或氧化铝材料优选包括介孔氧化硅材料;所述介孔氧化硅材料平均孔径优选为2.5~25nm。技术方案4.根据技术方案1~3任一所述的方法,其中,所述的镁组分处于介孔氧化硅材料和/或氧化铝材料中形成含镁介孔含氧化硅材料和/或含镁氧化铝材料;优选的,含镁介孔含氧化硅材料中mgo与sio2的重量比值为0.5~30:70~99.5例如为2~30:70~98或5~25:75~95,含镁氧化铝材料中mgo与al2o3的重量比值为0.5~30:70~99.5例如为2~30:70~98或5~25:75~95;优选的,所述的活性组分处于含镁介孔氧化硅材料和/或含镁氧化铝材料中。技术方案5.根据技术方案1~4任一所述的方法,其中,所述的no氧化催化剂包括粘结剂和任选的粘土。所述的粘结剂例如氧化铝粘结剂、氧化锆粘结剂、氧化钛粘结剂、氧化硅-氧化铝粘结剂中的一种或多种。所述的氧化铝粘结剂例如拟薄水铝石和/或铝溶胶;所述的氧化锆粘结剂例如锆溶胶和/或锆凝胶,所述的氧化钛粘结剂例如钛溶胶和或/钛凝胶,所述的氧化硅-氧化铝粘结剂例如硅铝溶胶和/或硅铝凝胶。技术方案6.根据技术方案1~5任一所述的方法,其中,所述的no氧化催化剂中含5~95重量%例如10~80重量%含镁介孔含氧化硅材料、0.5~40重量%例如5~40重量%或10~30重量%的活性金属氧化物、5~50重量%例如5~30重量%或5~20重量%的粘结剂和1-50重量%例如2~30重量%或5~20重量%粘土。技术方案7.根据技术方案1~6任一所述的方法,其中,所述no氧化催化剂包括:氧化活性组分和载体材料;所述载体材料含有二氧化硅和镁元素,所述载体材料具有介孔结构,所述载体材料的比表面积为300m2/g以上,平均孔径为2.5~25nm。技术方案8.根据技术方案7所述的方法,其中,所述载体材料的比表面积为400~800m2/g,平均孔径为6~20nm。技术方案9.根据技术方案7或8所述的方法,其中,所述载体材料的xrd谱图在2θ角为0.1°至2.5°处优选0.8°至1.4°例如0.9°~1.3°处存在衍射峰;优选的,还在15°至25°处存在衍射峰。技术方案10.根据技术方案7~9任一所述的方法,其中,所述载体材料中,以氧化镁计的镁元素的重量含量为0.5%~30%例如为5~25重量%或10~20重量%或10~15重量%;通常的,氧化硅含量为70~99.5重量%例如为75~95重量%或80~90重量%或85~90重量%。技术方案11.根据技术方案7~10任一所述的的方法,其中,所述载体材料中,包括掺杂的镁元素和浸渍的镁元素,以镁元素的总重量为基准,所述载体材料中掺杂的镁元素占3%-50%;浸渍的镁元素占50-97%。技术方案12.根据技术方案7~11任一所述的的方法,其中,以所述no氧化催化剂的重量为基准,按照干基计,所述no氧化催化剂含有0.4~40重量%例如5~40重量%的氧化活性组分、5~95重量%例如60-90重量%的所述载体材料、1~50重量%的粘土和5~50重量%例如7~40重量%或10~30重量%的粘结剂。所述的氧化活性组分选自ivb族、vb族、vib族、viib族、viii族、ib和iib族金属或它们的氧化物中的一种或多种。技术方案13.根据技术方案1~12任一项所述的方法,其中,于一实施方案,所述的氧化活性组分包括贵金属和任选的其它金属(所述其它金属是指其它活性金属)氧化物,所述贵金属选自pt、pd、ru、rh、os、ir中的一种或多种,所述的其它金属氧化物活性组分包括vib族、viib族、iib族和viii族非贵金属氧化物中的一种或多种,其中粘结剂可以是氧化铝、ⅳb族氧化物中的一种或多种;该组合物可以具有较高的氧化活性;优选的,以no氧化催化剂的重量为基准,no氧化催化剂中贵金属的含量为0.4~10重量%例如0.5~2重量%,其它金属氧化物的含量为0~39.6重量%;于一实施方案,所述的氧化活性组分包括vib和/或viib族金属氧化物例如mn氧化物和/或cr氧化物以及任选其它金属氧化物例如fe、co、ni、cu氧化物中的一种或多种;其中粘结剂可以是氧化铝、ⅳb族氧化物(氧化钛和/或氧化锆)中的一种或多种;该组合物可以具有较低的成本,并具有较高的氧化活性,可以具有较高的低温氧化活性;优选的,以no氧化催化剂的重量为基准,no氧化催化剂中vib和/或viib族金属氧化物的含量为2~40重量%例如5~20重量%,其它金属氧化物的含量为0~38重量%,所述其它金属氧化物优选为fe、co、ni、cu氧化物中的一种或多种;于一实施方案,所述的氧化活性组分包括ib族金属氧化物例如铜氧化物,以及任选其它金属氧化物例如fe、co、ni的氧化物中的一种或多种,其中粘结剂可以是氧化铝、ⅳb族氧化物中的一种或多种,该组合物可以具有较低的再生温度。优选的,以no氧化催化剂的重量为基准,no氧化催化剂中ib族金属氧化物的含量为1~30重量%例如5~25重量%或10~15重量%,其它金属氧化物的含量为0~39重量%,所述其它金属氧化物优选为fe、co、ni氧化物中的一种或多种。其中,所述的氧化铝粘结剂可以为焙烧后得到三氧化二铝的粘结剂,例如为酸化拟薄水铝石、酸化sb粉或铝溶胶,或者为他们中两者或三者的组合。其中酸化过程具体为,用酸与sb粉或者拟薄水铝石反应,反应温度为室温~95℃,例如15~95℃,反应时间为0.5~8h,使用的酸可以包括盐酸、磷酸、草酸、硝酸中的一种或者多种。所述的ⅳb族氧化物粘结剂可以为焙烧后得到iv族元素氧化物的粘结剂,例如为含ti粘结剂和/或含zr粘结剂,以进一步提高催化剂的no催化氧化性能;优选地,粘结剂可以为酸化二氧化锆、四氯化钛、钛酸乙酯、钛酸异丙酯、醋酸钛、水合氧化钛、锐钛矿型二氧化钛、四氯化锆、氢氧化锆、醋酸锆、水合氧化锆和无定形二氧化锆,或者为他们中两者或三者的组合。其中酸化二氧化锆的酸化过程可以包括:二氧化锆与去离子水打浆,然后进行酸化,酸化的酸可以是盐酸、硝酸、草酸、磷酸、中的一种或者多种。技术方案14.根据技术方案1~13任一所述的方法,其中,所述的no氧化催化剂中镁组分和氧化活性组分处于相同的颗粒中,或处于不同的颗粒中;当镁组分和氧化活性组分处于不同的颗粒中时,可以使含镁组分的催化剂组合物颗粒和含氧化活性组分的催化剂组合物颗粒按比例混合形成no氧化催化剂。技术方案15.根据技术方案1~14任一所述的方法,其中,还包括将所述的no氧化催化剂再生的步骤;所述再生可以采用氢气、一氧化碳、气体烃中的一种或多种进行还原;再生产物可以引入克劳斯制硫过程。技术方案16.根据技术方案1~15任一所述的方法,其中,所述的含硫氧化物、氧气和no的气体为fcc再生烟气、热电厂尾气、锅炉产生的废气、燃烧炉产生的废气或煅烧炉产生的废气。优选的,所述含硫氧化物、氧气和no的气体中氧气的含量不低于0.5体积%例如为1~10体积%。技术方案17.根据技术方案1~16任一所述的方法,其中,所述的含硫氧化物、氧气和no的气体是将不含氧气或氧气含量较低的含no和硫氧化物的气体通过引入氧气或空气得到的。技术方案18.根据技术方案1~17任一所述的方法,其中,所述的含硫氧化物、氧气和no的气体为催化裂化再生烟气,所述催化裂化再生烟气可以是完全再生过程产生的烟气或不完全再生过程产生的烟气;例如,所述完全再生过程烟气可以是三级旋风分离器后至余热锅炉之前的烟气,所述不完全再生过程烟气可以是co锅炉之后至脱硫设施之前的烟气。技术方案19.根据技术方案16、17或18所述的方法,其中,所述的含硫氧化物、氧气和no的气体中,sox的浓度为20-10000mg/m3,no浓度为20-2000mg/m3,氧气的浓度为不低于0.2体积%;优选的,所述的含硫氧化物、氧气和no的气体中氧气含量为0.5~8体积%;通常的,所述的含硫氧化物、氧气和no的气体中nox含量≥100mg/m3,sox含量大于0例如为20~5000mg/m3,一氧化氮(no)含量大于80mg/m3。技术方案20.根据技术方案1~19任一所述的方法,其中,所述接触反应的反应温度为250℃至450℃,例如为300℃至400℃。技术方案21.根据技术方案1~20任一所述的方法,其中,所述接触反应的反应压力为0-1mpa(表压)例如为0.1-0.5mpa。技术方案22.根据技术方案1~21任一项所述的方法,其中,所述接触反应在流化床反应器中进行。技术方案23.根据技术方案22所述的方法,其中,所述流化床反应的质量空速为1-100h-1或5-50h-1或5-20h-1。技术方案24.根据技术方案22~23任一所述的方法,其中,所述no氧化催化剂为微球颗粒状,其平均直径粒径为60~80微米,颗粒直径不超过149微米的颗粒含量不低于90体积%。技术方案25.根据技术方案1~24任一所述的方法,其中,还包括将no氧化催化剂再生的步骤,例如可以将no氧化流化床反应器中的no氧化催化剂引入流化床再生器再生;也可以是多个no氧化反应器并联操作,部分no氧化反应器进行所述no氧化反应,部分反应器进行no氧化催化剂再生。技术方案26.一种含硫氧化物、氧气和no的烟气的处理方法,包括,按照技术方案1~25任一项所述的方法将含硫氧化物、氧气和no的烟气与no氧化催化剂接触反应使no氧化,然后脱除烟气中的nox和sox。可得到nox和sox含量符合国家排放标准要求的烟气。技术方案27.根据技术方案26所述的方法,其中,所述脱除烟气中的nox和sox的方法可采用吸收的方法脱除;吸收的方法可以采用干法或湿法,湿法吸收可采用现有烟气洗涤方法和系统,例如中国专利申请201810491397.7、201820761232.2、201820761234.1或201820761233.7提供的洗涤方法或烟气洗涤系统。技术方案28.一种含硫氧化物、氧气和no气体氧化处理系统,包括流化床氧化反应器和no氧化催化剂再生器,所述流化床氧化反应器中装有颗粒状含镁的no氧化催化剂,用于将含sox、nox和氧气的气体中的no氧化,no氧化催化剂再生器用于将no氧化催化剂再生。技术方案29.根据技术方案28所述的系统,其中,所述流化床反应器上设置旋风分离装置,以将破损no氧化催化剂的细小颗粒导出流化床反应器。技术方案30.根据技术方案28或29所述的系统,其中,所述系统还包含催化组合物藏量测量及加剂装置,以便随时对催化组合物进行补充和置换。技术方案31.根据技术方案28~30任一所述的系统,其中,还包括流化床再生器,流化床再生器用于使与镁结合的硫氧化物转变为气体。本发明提供的no氧化方法,可以在sox存在情况下,具有较高的no转化率,此外可以长时间维持较高的no转化率。失活后的催化剂可通过再生,恢复活性。本发明提供的方法可用于含硫氧化物烟气脱氮。附图说明图1为本发明提供的一种实施方式的流程示意图,其中1为产生含no的烟气的装置,2为no氧化反应器,a为含氧气的气体入口,b为含no的烟气出口,c为no氧化反应后的烟气出口,d再生后的气体出口,e为再生气体入口。图2为本发明提供的第二种实施方式的流程示意图,其中1为产生含no的烟气的装置,2为no氧化反应器,3为no氧化催化剂再生器,4为待生催化剂输送管线,5为再生催化剂输送管线,a为含氧气的气体入口,b为含no的烟气出口,c为no氧化反应后的烟气出口,d为再生气体出口,e为再生气体入口。具体实施方式本发明提供的含硫氧化物、氧气和no的气体中no的氧化方法,其中所述的no氧化催化剂可以按照现有方法制备,例如形成含氧化活性组分和氧化镁组分的混合物、成型、焙烧或者将含氧化镁组分的物质成型,浸渍引入氧化活性组分、焙烧。一种实施方式,所述的no氧化催化剂包括镁组分、氧化活性组分和基质,可以通过形成镁组分、氧化活性组分和基质的混合物、成型、焙烧得到,也可以通过形成包括部分或全部基质和氧化镁组分的混合物、浸渍引入氧化活性组分然后与其余基质混合、成型、焙烧得到。其中焙烧前还可以包括干燥的步骤。以所述no氧化催化剂的干基重量为基准,所述的活性组分的含量为0.4~40重量%例如5~40重量%,镁组分的含量为0.3-28.5重量%,基质含量为5~95重量%。干基重量,是指物质在800℃焙烧1小时后的固体产物重量。本发明提供的含硫氧化物、氧气和no的气体中no的氧化方法,其中所述的no氧化催化剂优选包括氧化活性组分和含镁和硅的载体材料简称载体材料,优选的,所述载体材料含有二氧化硅和镁元素,所述载体材料具有介孔结构,所述载体材料的比表面积为300m2/g以上,平均孔径为2.5~25nm。所述载体材料,可通过包括以下步骤的方法制备:a、使硅源、结构导向剂、水与任选的第一镁源接触反应,并使反应所得产物进行第一焙烧,得到介孔含氧化硅材料;任选的,b、在第一浸渍条件下,使含有第二镁源的第一浸渍液与所述介孔含氧化硅材料接触进行第一浸渍,任选进行干燥和/或第二焙烧,得到所述载体材料;其中a和b至少一个步骤中与镁源接触引入镁元素。优选的,步骤a中,硅源以sio2计、结构导向剂、水与任选的第一镁源的摩尔比优选为1:(0.25~7):(2~40):(0~0.319)。当步骤a中引入第一镁源的情况下,第一镁源与硅源的摩尔比优选为(0.0037~0.22):1;当步骤b中引入第二镁源的情况下,第二镁源以mgo计与所述介孔含氧化硅材料的重量比为0.002~0.41:1,优选为0.005~0.29:1。所述载体材料制备方法中,步骤a中含有硅源和结构导向剂的反应原料与任选的第一镁源接触反应,反应温度为150~200℃,反应时间10~72h例如24~60h。优选的,所述使硅源、结构导向剂与任选的第一镁源接触反应包括:将所述硅源、所述结构导向剂、水与所述第一镁源混合,将得到的混合液熟化,升温反应形成凝胶;使所述凝胶在反应釜内在150~200℃下反应10~72h;将反应所得产物进行所述第一焙烧后得到所述介孔含氧化硅材料。步骤a中可以引入或不引入镁元素,优选情况下,以镁元素计,所述第一镁源的用量为镁元素总用量的3-50重量%。所述载体材料制备方法中,步骤a中,优选情况下以氧化硅计的所述硅源与以氧化镁计的所述第一镁源的用量重量比为1:(0~0.21)例如1:(0.00015~0.15);所述第一焙烧的条件包括:在含氧气氛下焙烧,焙烧温度为500℃~800℃,焙烧时间为8~20h。所述硅源选可自硅溶胶、水玻璃和有机硅酯中的至少一种;所述结构导向剂可选自醇胺、有机季铵化合物、有机胺、环烷基砜和多元醇中的至少一种,所述有机硅酯例如硅酸四甲酯、硅酸四乙酯、硅酸四丁酯、二甲基二乙基硅酯中的一种或多种。所述醇胺例如为三乙醇胺,所述有机季铵化合物例如为四乙基氢氧化铵,所述环烷基砜例如为环丁砜,所述有机胺为例如四乙基五胺,所述多元醇例如为乙二醇、丙三醇、二甘醇、三甘醇和四甘醇中的至少一种。所述硅源优选为有机硅酯(有机硅源)例如硅酸四乙酯、所述结构导向剂优选为三乙醇胺和可选的四乙基氢氧化铵,步骤a所述方法包括:使所述硅源、三乙醇胺、四乙基氢氧化铵、水和以氧化镁计的所述第一镁源按照1:(0.25~2):(0~6):(2~40):(0~0.319)的摩尔比混合,将得到的混合液在10~40℃下熟化6~24h,并在空气气氛中在40~120℃下反应12~24h,形成凝胶,以sio2计的硅源与以氧化镁计的所述第一镁源的摩尔比优选1:0.0037~0.22;然后使所述凝胶在反应釜内在150~200℃下反应10~72h,将反应所得产物在空气气氛下500-800℃进行所述第一焙烧,焙烧时间优选8~20h,得到所述介孔含氧化硅材料。优选的,所述第一焙烧方法如下:使所述反应所得产物在空气气氛下以每分钟0.05~2℃的速率升温至500~800℃进行所述第一焙烧。所述载体材料制备方法中,在第一浸渍条件下,使含有第二镁源的第一浸渍液与所述介孔含氧化硅材料接触进行第一浸渍,然后经干燥和/或第二焙烧后得到所述载体材料。当仅在步骤a中引入镁组分的时候,所述方法不包括步骤b,步骤a得到的介孔含氧化硅材料可以直接作为载体材料使用。其中,步骤b中,所述第一浸渍包括:将所述第二镁源溶解在水中或者将所述第二镁源与水打浆,得到所述第一浸渍液;使所述第一浸渍液与所述介孔含氧化硅材料进行等体积浸渍;优选的,所述第一浸渍的条件包括:浸渍温度为10~80℃,时间为1~24h,所述第一浸渍液中以氧化物计的镁和以干基计的所述介孔含氧化硅材料的重量比为(0.002~0.41):1例如0.005~0.29:1。所述第一镁源和所述第二镁源各自独立地为硝酸镁、氢氧化镁、醋酸镁、碳酸镁或氯化镁,或者为他们中两者或三者或四者的组合。本发明提供的含硫氧化物、氧气和no的气体中no氧化方法中,以所述no氧化催化剂的总重量为基准,所述no氧化催化剂含有0.4~40重量%例如5~40重量%的氧化活性组分(也称活性金属组分)和5~95重量%例如10~90%或15~85重量%或20-80重量%的所述载体材料。所述氧化活性组分例如过渡金属和/或过渡金属氧化物,过渡金属元素(活性金属)可以选自iiib族、ivb族、vb族、vib族、viib族、viii族、ib族和iib族金属元素中的一种或几种,例如过渡金属元素可以为sc、y、ti、zr、hf、v、nb、ta、cr、mo、w、mn、tc、re、fe、ru、os、co、rh、ir、ni、pd、pt、cu、ag、au、zn、cd中的一种或多种例如为其中的一种或者为他们中两者或三者或多种的组合;在一种具体实施方式中,所述活性金属为iiib族元素、ivb族元素、vb族元素和iib族元素中的一种或几种,例如为sc、y、ti、zr、hf、v、nb、ta、cr、mo、w、zn、cd中的一种,或者为他们中两者或三者或多种的组合例如为ti、v和zr中的至少一种。所述活性金属氧化物的重量含量为0.4%~40%例如5~30重量%或10~25重量%。在本发明提供的no氧化催化剂中,活性金属可以以氧化物形式存在,氧化物的重量含量可以在较大范围变化,为了进一步提供适宜的催化氧化能力,活性金属氧化物的重量含量优选为10%~25%,例如为12%~20%、15%~22%或14~18%。在根据本公开的催化剂中,载体材料的重量含量可以5%~95%,优选为40%~80%,例如为42%~78%、45%~70%或50%~75%。其中,为了进一步促进活性金属在载体上分散,优选地,载体材料中,以载体材料的总重量为基准,氧化镁的重量含量可以为0.5%~30%,优选为5%~20%,例如为6%~18%或7.2%~15%。本发明提供的含硫氧化物、氧气和no的气体中no氧化方法中,于一种实施方式,所述的氧化活性组分包括贵金属和任选的其它金属氧化物,所述贵金属选自pt、pd、ru、rh、os、ir中的一种或多种,所述的其它金属氧化物活性组分包括vib族、viib族、iib族和viii族非贵金属氧化物中的一种或多种,其中粘结剂可以是氧化铝、ⅳb族氧化物中的一种或多种;该组合物可以具有较高的氧化活性。优选的,以no氧化催化剂的重量为基准,所述no氧化催化剂中贵金属的含量为0.4~10重量%例如0.5~2重量%,其它金属氧化物的含量为0~39.6重量%,所述载体材料含量为5~95重量%例如10-90重量%或60-90重量%或65~85重量%,粘土含量为1~50重量%例如5-20重量%,粘结剂含量为5~50重量%例如7~40重量%或5~30重量%或10~30重量%。于一实施方式,所述的氧化活性组分包括mn氧化物和/或cr氧化物以及任选fe、co、ni、cu氧化物中的一种或多种;其中粘结剂可以是氧化铝、ⅳb族氧化物(氧化钛和/或氧化锆)中的一种或多种;该组合物可以具有较低的成本,并具有较高的氧化活性;优选的,以no氧化催化剂的重量为基准,no氧化催化剂中mn氧化物和/或cr氧化物的含量为2~40重量%例如5~20重量%,其它金属氧化物的含量为0~38重量%,所述其它金属氧化物优选为fe、co、ni、cu氧化物中的一种或多种,所述载体材料含量为5~95重量%例如10~90重量%或60-90重量%或60~95重量%或65~85重量%,粘土含量为1~50重量%例如5-20重量%,粘结剂含量为5~50重量%例如7~40重量%或10~30重量%或5~30重量%。于一实施方式,所述的氧化活性组分包括铜氧化物,以及任选fe、co、ni的氧化物,其中粘结剂可以是氧化铝、ⅳb族氧化物中的一种或多种,该组合物可以具有较低的起燃温度。优选的,以no氧化催化剂的重量为基准,no氧化催化剂中cu氧化物的含量为1~30重量%例如5~25重量%或10~15重量%,其它金属氧化物的含量为0~39重量%,所述其它金属氧化物优选为fe、co、ni氧化物中的一种或多种,所述载体材料含量为5~95重量%例如10~90重量%或65-90重量%或60~90重量%,粘土含量为1~50重量%或5~20重量%,粘结剂含量为5~50重量%例如7~40重量%或10~30重量%或5~30重量%。所述的no氧化催化剂中,还可以包括粘土,粘土可以为本领域常规种类,例如为高岭土、海泡石土、凹凸棒、累脱土、蒙脱土或硅藻土,或者为他们中两者或三者或四者以上的组合。所述的no氧化催化剂中,还可以包括粘结剂,粘结剂可以为本领域常规种类,优选为氧化铝粘结剂、氧化锆粘结剂或氧化钛粘结剂,或者为它们中两者或三者的组合。本发明所述no氧化催化剂可以采用本领域常规方法制备,例如可以将氧化活性组分例如所述过渡金属元素(也称活性金属)通过浸渍法负载在本公开的载体材料上,然后与粘结剂、粘土混合打浆,再经喷雾干燥和第二焙烧后得到催化剂。在一种具体实施方式中,可以通过包括如下步骤的方法制备本发明所述的no氧化催化剂:c、在第二浸渍条件下,使含有活性金属前体的第二浸渍液与所述载体材料接触进行第二浸渍,得到浸渍有活性金属的载体材料;d、使含铝粘结剂、粘土与浸渍有活性金属的载体材料进行混合打浆,然后经喷雾干燥和第三焙烧后得到催化剂;其中,活性金属前体含有过渡金属元素中的至少一种。本发明所述no氧化催化剂制备方法中,步骤c的第二浸渍可以为本领域常规的方法和条件,例如在一种实施方式中,第二浸渍可以包括:使载体材料与含有活性金属前体的第二浸渍液混合均匀后在10~40℃下静置1~24h,优选在15~30℃下静置10~24h;第二浸渍液中水和以干基重量计的载体材料的重量比可以为(0.58~1.2):1,例如为(0.7~1.1):1或(0.6~0.9):1。以氧化物计的活性金属:以干基计的载体材料的重量比为0.004~0.42:1例如为(0.083~0.42):1或(0.1~0.4):1。本发明所述no氧化催化剂制备方法中,活性金属前体含有所述的过渡金属元素,活性金属前体可以包括活性金属硝酸盐、活性金属碳酸盐、活性金属醋酸络合物、活性金属氢氧化物、活性金属草酸盐络合物和活性金属酸盐中的一种或多种,优选为活性金属硝酸盐和/或高价活性金属酸盐,这些过渡金属的盐为本领域技术人员所熟知,本发明不再赘述。本发明所述no氧化催化剂制备方法中,步骤d中,所述第三焙烧的条件可以包括:在空气气氛下焙烧,焙烧温度可以为250℃~800℃,优选350℃~700℃,更优选350℃~450℃,焙烧时间可以为1~12h,优选为4~10h。本发明所述no氧化催化剂制备方法中,步骤d中,含铝粘结剂、粘土与吸附活性金属的载体材料的用量可以在较大范围内变化,优选地,以氧化铝计的含铝粘结剂、粘土与以干基计的浸渍有活性金属的载体材料的用量重量比可以为1:(0.002~10):(1.3~27),优选为1:(0.5~8):(5~20)。本发明所述no氧化催化剂制备方法中,粘结剂为包括焙烧后得到三氧化二铝的粘结剂,例如为酸化拟薄水铝石、酸化sb粉或铝溶胶,或者为他们中两者或三者的组合。其中酸化过程具体为,用酸与sb粉或者拟薄水铝石反应,反应温度为室温~95℃,例如15~95℃,反应时间为0.5~8h,使用的酸可以包括盐酸、磷酸、草酸、硝酸中的一种或者多种。在另一种实施方式中,粘结剂为包括焙烧后得到ivb族元素氧化物的粘结剂,例如为含ti粘结剂和/或含zr粘结剂,以进一步提高催化剂的no催化氧化性能。优选地,所述焙烧后得到ivb族元素氧化物的粘结剂可以为酸化二氧化锆、四氯化钛、钛酸乙酯、钛酸异丙酯、醋酸钛、水合氧化钛、锐钛矿型二氧化钛、四氯化锆、氢氧化锆、醋酸锆、水合氧化锆和无定形二氧化锆,或者为他们中两者或三者的组合。其中酸化二氧化锆的酸化过程可以包括:二氧化锆与去离子水打浆,然后进行酸化,酸化的酸可以是盐酸、硝酸、草酸、磷酸、中的一种或者多种。本发明所述的no氧化催化剂制备方法中,浸渍活性金属的载体材料与粘土和无机粘结剂混合打浆前可以进行干燥也可以不进行干燥。所述的喷雾干燥的方法为本领域技术人员熟知,本发明没有特殊要求。下面的实施例对本发明进一步说明,但并不用于限制本发明:拟薄水铝石为山东铝厂产品,sb粉为aldrich公司产品,正硅酸乙酯(teos)购买于aldrich公司,三乙醇胺(tea)购买于fluka,四乙基氢氧化铵(teaoh)购买于aldrich公司。高硅铝比zsm-5分子筛购买于齐鲁华信公司,硅铝原子比为170,命名为zsm-5-170,比表面积为348m2/g;比表面为50m2/g的sio2购自赢创德固赛(中国)投资有限公司。对比例和实施例中所用化学试剂未特别注明的,其规格为化学纯。铝溶胶,中国石化催化剂有限公司齐鲁分公司产品,al2o3含量为21.3重量%。在各实施例中载体材料的比表面积、孔体积、平均孔径由低温氮吸附-脱附法测定(bet法),bet比表面和孔体积的测试方法采用氮吸附容量法,按照bjh计算方法。(参见石油化工分析方法(ripp试验方法),ripp151-90)实施例1在剧烈搅拌的条件下,将216g的三乙醇胺(tea)、25.56g醋酸镁和54g的去离子水滴加到300g的teos中,反应40min,得到第一混合物,再将300g的teaoh滴加到上述第一混合物中得到第二混合物。第二混合物在30℃下熟化24h,然后在空气气氛中98℃加热24h得到凝胶。将该凝胶置于反应釜内,在180℃下反应16h。最后,将产物在空气气氛中以每分钟1℃的速率升温到600℃焙烧10h,得到焙烧产物。将30.56g六水硝酸镁溶于86.5g去离子水中,等体积浸渍于焙烧产物上,室温放置熟化24h,然后空气中100℃干燥12h,500℃焙烧4h,得到本实施例的载体材料,记为介孔分子筛载体材料a。载体材料a的比表面为455m2/g,平均孔径为8.1nm;载体材料a的xrd谱图在2θ为0.89°和22.68°处分别有衍射峰。将钛酸丁酯溶解到去离子水中得到钛酸丁酯水溶液,钛酸丁酯:水重量比为4.259:8,按照水:载体材料a=1:1的重量比将钛酸丁酯水溶液加入载体材料a中,室温下静置5h,得到浸渍有活性金属的载体材料。将铝溶胶与去离子水混合,得到第一混合液,搅拌10min,其中以干基计铝溶胶与水的重量比为1:6;按照以干基计的高岭土:以干基计的铝溶胶=1:1的重量比将高岭土与第一混合液打浆,搅拌60min得到第二混合液;将第二混合液与浸渍有活性金属的载体材料进行混合打浆,以干基计第二混合液与浸渍有活性金属的载体材料重量比为1:9,打浆时间30min,得到第三浆液。将第三浆液进行喷雾干燥、450℃下焙烧4h,得到催化剂cat-1。实施例2将284.16g的teos、18.39g六水硝酸镁与136.87g的去离子水混合得到第一混合物。在剧烈搅拌的条件下,将52.11g的tea以每分钟4~6g的速度滴加到第一混合物中,得到第二混合物。将第二混合物在25℃下熟化16h,然后在空气气氛中99℃加热24h得到凝胶。将该凝胶置于反应釜内,在190℃下反应48h。最后,将产物在空气中以每分钟1℃的速率升温到550℃焙烧10h,得到焙烧产物。将61.53g醋酸镁溶于81.9g去离子水中,等体积浸渍于焙烧产物上,室温放置熟化18h,然后空气中120℃干燥18h,450℃焙烧5h,得到本实施例的载体材料,记为载体材料b。载体材料b分子筛的比表面为568m2/g,平均孔径为7.0nm;载体材料b的xrd谱图在2θ为1.23°和23.35°处分别有衍射峰。参考实施例1的方法,用载体材料b代替载体材料a,制备催化剂cat-2。实施例3将173.7g的tea,5.86g六水硝酸镁和569.8g的去离子混合,得到第一混合物,在剧烈搅拌的条件下,将255.5g的teos滴加到第一混合物中,得到第二混合物,将第二混合物在40℃下熟化24h,然后在空气气氛中100℃加热18h得到凝胶。将该凝胶置于反应釜内,在170℃下反应48h。该胶体在空气气氛中以每分钟1℃的速率升温到550℃,在550℃焙烧10h,得到焙烧产物。将98.90g六水硝酸镁溶于73.6g去离子水中,等体积浸渍于焙烧产物上,室温放置熟化15h,然后空气中120℃干燥18h,520℃焙烧3h,得到本实施例的载体材料,记为载体材料c。载体材料c分子筛的比表面为443m2/g,平均孔径为18.2nm;载体材料c的xrd谱图在2θ为1.19°和22.13°处分别有衍射峰。参考实施例1的方法,使用载体材料c,制备催化剂cat-3。对比例1将173.7的tea,569.8g的去离子混合,得到第一混合物,在剧烈搅拌的条件下,将255.5g的teos滴加到第一混合物得到第二混合物,将第二混合物在40℃下熟化24h,然后在空气气氛中100℃加热18h得到凝胶。将该凝胶置于反应釜内,在170℃下反应48h。该胶体在空气气氛中以每分钟1℃的速率升温到550℃焙烧10h,得到焙烧产物,记为载体材料d。载体材料d材料的比表面积为471m2/g,平均孔径为19.7nm;载体材料d的xrd谱图在2θ为0.98°和23.08°处分别有衍射峰。参考实施例3的方法,使用载体材料d代替载体材料c,制备催化剂dcat-1.实施例4参考实施例1的方法,改变组分和配比,使用载体材料a制备催化剂,记为cat-4。实施例5参考实施例1的方法,改变化学组成,使用载体材料b制备催化剂,记为cat-5。实施例6载体材料制备过程与实施例1的不同在于,凝胶在反应釜中反应2h;得到的载体材料g材料的比表面积为873m2/g,平均孔径为3.0nm;以镁元素的总重计;载体材料g的xrd谱图在2θ为0.84°和22.34°处分别有衍射峰。参考实施例1的方法制备催化剂cat-6。实施例7载体材料制备过程与实施例1的不同在于,在第一混合物中不加入醋酸镁,而在浸渍步骤中增加等当量的硝酸镁,得到的载体材料e材料的比表面为积469m2/g,平均孔径为9.8nm;以镁元素的总重计,载体材料e分子筛的不含掺杂镁,表面和/或孔中含有100%的镁;载体材料e分子筛的xrd谱图在2θ为1.12°和22.64°处分别有衍射峰。参考实施例1的方法制备催化剂cat-7。制备例8载体材料制备过程与实施例1的不同在于,不进行六水硝酸镁浸渍的步骤,而在第一混合物中相应增加等当量的醋酸镁;得到的载体材料f材料的比表面积为470m2/g,平均孔径为10.1nm;以镁元素的总重计,载体材料f分子筛的骨架含有100%的镁,表面和/或孔中不含镁;载体材料f分子筛的xrd谱图在2θ为1.14°和23.38°处分别有衍射峰。参考实施例1的方法制备催化剂cat-8。对比例2将300gsb粉在空气气氛中450℃焙烧4h,得到γ-al2o3载体,记为γ-al2o3-a。γ-al2o3-a载体的比表面积为233m2/g,平均孔径为7.5nm,xrd谱图在2θ为0.1°~2.5°处无衍射峰。按照实施例3的方法,使用γ-al2o3-a载体制备催化剂dcat-2实施例9用硝酸镁溶液浸渍γ-al2o3-a,550℃焙烧1h,得到载体γ-al2o3-a-mg,然后按照实施例3的方法,用载体γ-al2o3-a-mg代替载体材料c制备催化剂cat-9。实施例10参考实施例1,使用载体材料a,活性组分采用重铬酸铵,使用钛溶胶粘结剂,制备催化剂cat-10。实施例11参考实施例1,采用载体a,使用硝酸铜,使用钛溶胶粘结剂,制备催化剂,记为cat-11。实施例12参考实施例1,采用载体a,使用氯铂酸铵,使用钛溶胶粘结剂制备催化剂。记为cat-12。载体材料a~g的组成见表1。催化剂cat-1~cat-12、dcat-1和dcat-2的投料配比见表2(其中的配比为重量比,以干基计)。表1表2表2续测试例用于说明含硫氧化物、氧气和no的气体中no的氧化方法。no氧化反应在固定床反应器中进行。具体实验条件见表3。混合气体中各组分采用傅立叶红外进行检测,检测温度为190℃,样品池体积为0.2l,光程5.11米。水蒸气气化炉温度240℃,汽化后水蒸气与模拟烟气混合,进行反应,反应后的混合气采用全程保温,以确保混合气中水蒸气不冷凝,测试结果准确。no氧化转化率:反应稳定15min后测量,具体计算方法为:转化率=(1-反应器出口混合气体中no浓度/反应器入口混合气体中no浓度)×100%;活性降低量(%)=烟气不含硫条件下测得的no氧化转化率-抗硫性能试验(烟气含硫)转化率其中,no氧化试验转化率为在表3所示的no氧化试验条件下测得的no转化率,抗硫性能试验转化率表3所示的抗硫性能试验条件下测得的no转化率。计算结果列于表4。其中,抗硫性能试验转化率是指在表3的抗硫性能试验条件下(烟气有含有硫氧化物)测试的转化率。表3表4催化剂反应温度(℃)no氧化试验转化率抗硫性能试验转化率活性降低量cat-130035.6%34.3%1.3%cat-235042.3%41.0%1.3%cat-335043.4%41.8%1.6%cat-440048.4%47.0%1.4%cat-540047.3%45.6%1.7%cat-630033.3%31.9%1.4%cat-730031.9%28.3%3.6%cat-830033.7%28.9%4.8%dcat-135042.8%18.2%24.6%cat-935035.3%21.4%13.9%dcat-235034.7%7.5%27.2%cat-1035050.3%48.7%1.6%cat-1130048.2%46.8%1.4%cat-1225087.5%85.9%1.6%由表4可见,本发明提供的方法,在no氧化催化剂活性组分含量相同的情况下,可以具有更高的no转化率。此外,本公开的各种不同的实施方式之间也可以进行任意组合,只要其不违背本公开的思想,其同样应当视为本公开所公开的内容。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1