一种光热协同增强全光谱响应异质结构光催化剂及其制备的制作方法
本发明属于半导体光催化剂制备技术领域,涉及一种异质结构光催化剂的制备方法,尤其是具体涉及一种光热协同增强全光谱响应异质结构光催化剂及其制备。
背景技术:
面对日益严重的水污染问题,利用绿色清洁的可再生的太阳能为驱动力的光降解技术是一种有效的应对办法。目前,大部分光催化剂(tio2、zno、cds、g-c3n4等)只对紫外光或者可见光有响应,无法充分利用太阳能,特别是占太阳能能量比重超过50%的近红外光,因此,近年来,全谱响应光催化剂引起人们的广泛研究兴趣,例如,果崇申课题组合成铯钨青铜csxwo3并研究了其全谱光催化降解有机染料mb的效率[appliedcatalysisb:environmental183(2016)142-148];yanglf课题组报道了铵钨青铜(nh4)xwo3与p25复合组成的光催化剂具有全谱光催化降解有机染料rhb性能[scientificreports7(2017)45715]。然而,无论是纯的钨青铜还是与p25复合的钨青铜在近红外区域的光催化效果不够理想,如铯钨青铜csxwo3在近红外光照射下光降解有机染料mb的去除率只有37%,光催化剂p25/(nh4)xwo3在近红外光照射下12小时只降解了60%的rhb。所以,寻找全光谱响应的高效光催化剂,探索新的方法并改善其全谱光催化效率成为当前研究的热点。
在研究全谱光催化中的近红外区域时,近红外光具有很强的热效应,同时光催化剂和有机废水在全谱光催化剂的实际应用中吸收太阳光时会不可避免地产生热能,改变反应温度,影响光催化进程,通过阿累尼乌斯公式kap=kap0exp[-(ea/rt)]得到温度越高表观速率越大。因此,研究温度对光催化的影响是必要的,这有利于光催化在降解有机废水上的实际应用。目前,已经有研究人员进行光热催化的探索。比如,behnajadyma[journalofhazardousmaterials133(2006)226-232]使用zno进行光催化降解c.i.酸黄23(ay23)时,讨论了温度对催化的影响;noureddinebarka等人[journalofphotochemistryandphotobiologya:chemistry195(2008)346–351]研究了使用无纺布包覆tio2作为光催化剂降解罗丹明b(rhb),探究温度从25℃到40℃对光催化的影响,结果表明光降解在较高温度下有较高的降解速率。目前普遍认为的机理是——温度对光催化反应的影响主要通过两方面:一是温度的变化会影响光催化剂对有机染料的吸附,二是温度的升高有助于提高反应的光生电子-空穴的有效复合,从而提高了去除有机染料的速率,达到更高的光催化效率。因此温度应会较好地提高光催化活性。
目前,钨青铜的主要合成方法分为气相法、液相法和固相法三种。气相法反应速度快、产物粒度分散均匀并且纯度较高,但是对实验条件及实验仪器要求较高,成本较贵,不能普遍推广。液相法中最常见的是水热/溶剂热法,工艺较成熟,设备要求相比于气相法要低,制备的产物结构缺陷小、均匀性好且可以制备出多种形貌的钨青铜,但其产物一般较少且可能制备时间较长,不利于工业化生产。固相法相对于其他两种方法操作简单,设备要求不高,产量高,可大量制备,但现有技术中尚缺少用固相法合成含有钾钨青铜的异质结构光催化剂的公开报道。
技术实现要素:
本发明所要解决的技术问题是:针对现有技术中用固相法合成含有钾钨青铜的异质结构光催化剂尚无公开报道的缺陷与不足,提供一种光热协同增强全光谱响应异质结构光催化剂及其制备方法。该异质结构光催化剂制备方法简单、快捷、高产率,且可以在紫外-可见-近红外光辐照下高效降解有机染料并呈现光热协同增强效应。
本发明采用如下技术方案,来实现发明目的。
首先,本发明公开了一种光热协同增强全光谱响应异质结构光催化剂。该催化剂的化学组成式为k0.2mxwo3,其中m=li或na,0.3≤x≤0.4,并含有钾钨青铜k0.2wo3相、碱金属钨酸盐li2w2o7相或na2w2o7相构成的异质结构。
进一步地,所述的k0.2wo3为jcpdsno.83-1331;所述的li2w2o7为jcpdsno.73-0171;所述的na2w2o7为jcpdsno.70-0860。
进一步地,所述的一种光热协同增强全光谱响应异质结构光催化剂,能够在紫外、可见和近红外光辐照下,光催化高效去除有机染料,并呈现光热协同增强效应。
其次,本发明还公开了一种光热协同增强全光谱响应异质结构光催化剂的制备方法。制备方法包括以下步骤:
s1、计算与称量:根据设计制备k0.2mxwo3催化剂的摩尔数,称取含有同样摩尔数钨元素的钨酸盐,并按照钨酸盐中钨元素与有机酸中羧基的摩尔比为1:3,称取相应量的有机酸;
s2、制备三氧化钨前驱体:将步骤s1中称取的反应物装入球磨机中,球磨0.5-6小时后,取出球磨产物,经洗涤、固液分离和干燥,即得三氧化钨前驱体;
s3、制备钾钨青铜k0.2wo3:按照钾与前驱体中钨的摩尔比为1:5,称取相应量的碳酸钾,与步骤s2得到的前驱体混合均匀,放入热处理炉,在还原气体气氛下,500~800℃保温0.5~6小时,冷却,即得k0.2wo3;
s4、制备异质结构光催化剂k0.2mxwo3:按照k0.2mxwo3催化剂中m与前驱体中钨的摩尔比为x:1,称取相应量的m的碳酸盐,与步骤s3得到的k0.2mxwo3混合均匀,放入热处理炉,在还原气体气氛下,500~800℃保温0.5~6小时,冷却,即得k0.2mxwo3。
进一步地,步骤s1中所述的钨酸盐为碱金属钨酸盐、仲钨酸铵或偏钨酸铵中的一种。
进一步地,步骤s1中所述的有机酸为柠檬酸、酒石酸或草酸中的一种。
进一步地,步骤s2中所述的球磨机为行星球磨机、搅拌磨、砂磨机或振动磨。
进一步地,步骤s3和s4中还原气体为氢气、氮氢混合气、一氧化碳的一种。
进一步地,步骤s4中所述的碳酸盐为碳酸锂或碳酸钠。
由上述方法所制备的光热协同增强全光谱响应异质结构光催化剂,能大幅度提高光催化剂的催化效率,可应用于有机染料水污染的快速降解处理。
有益效果:
(1)本发明采用机械化学法合成三氧化钨前驱体,再按比例将前驱体与钾盐混合在还原气氛下退火得到钾钨青铜,再将其按比例与锂盐或钠盐混合退火制备得到一种新型的光热协同增强并具有全谱光响应范围的光催化剂,即k0.2mxwo3(m=li,na),钾钨青铜与m2w2o7(m=li,na)复合形成了异质结构,大幅度提高光催化剂催化效率,在处理有机染料水污染方面具有广阔的应用前景。
(2)本发明的制备方法,方法简单,制备方便,对设备要求低,无需分散剂,重复利用率高,绿色环保高效。
附图说明
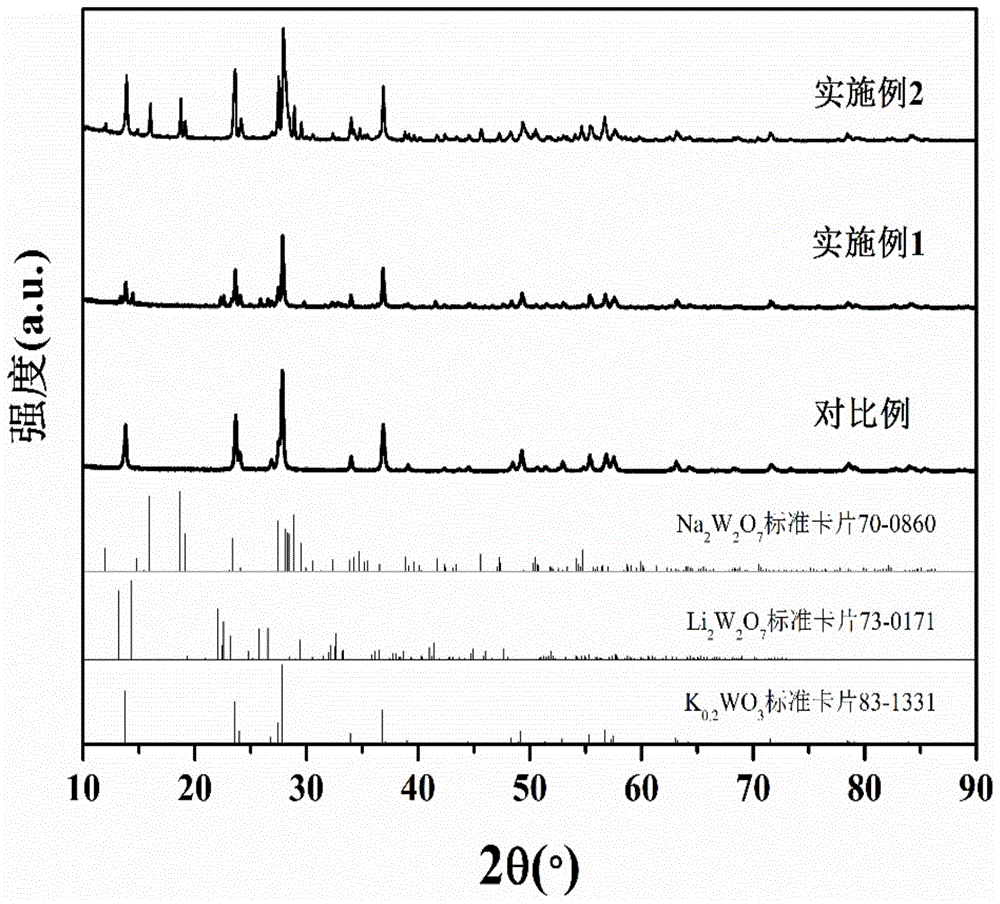
图1为对比例、实施例1和实施例2样品的x射线衍射图谱。
图2为对比例、实施例1和实施例2样品的扫描电镜照片。其中:图(a)为对比例样品,图(b)为实施例1样品,图(c)为实施例2样品。
图3为实施例1样品的透射电镜(a)和相应位置的高分辨率透射电镜(b)照片。
图4为实施例2样品的透射电镜(a)和高分辨率透射电镜(b)照片。
图5为30℃时对比例、实施例1和实施例2样品分别在紫外光(a)、可见光(b)和近红外光(c)辐照下去除溶液中rhb的变化曲线及其一级动力学速率常数柱形图(d)。
图6为不同温度时实施例1样品分别在紫外光(a)、可见光(b)和近红外光(c)辐照下去除溶液中rhb的变化曲线及其一级动力学速率常数柱形图(d)。
图7为不同温度时实施例2样品分别在紫外光(a)、可见光(b)和近红外光(c)辐照下去除溶液中rhb的变化曲线及其一级动力学速率常数柱形图(d)。
具体实施方式
下面结合具体实施例对本发明作进一步的说明,但本发明并不限于以下实施例。所述方法如无特别说明均为常规方法。所述原材料如无特别说明均能从公开商业途径获得。
实施例1:异质结构光催化剂k0.2li0.3wo3样品1的制备
称取8.246克二水合钨酸钠和1.576克二水合草酸,放入行星球磨机的球磨罐中以1800rpm的转速球磨3小时,取出产物,分别用去离子水洗3遍、乙醇洗3遍,离心分离得到的产物,放入干燥箱中80℃干燥4小时,即得前驱体。将所得前驱体和0.346克碳酸钾在研钵中混合均匀,放入通有h2/n2(体积:体积:5%/95%)混合气体的管式炉中,550℃保温1小时,冷却即得k0.2wo3。
将所得k0.2wo3与0.277克碳酸锂在研钵中混合均匀,放入通有h2/n2(体积:5%/95%)混合气体的管式炉中,550℃保温1小时,冷却,即可得k0.2li0.3wo3样品。
实施例2:异质结构光催化剂k0.2li0.3wo3样品2的制备
称取8.246克二水合钨酸钠和1.576克二水合草酸,放入行星球磨机的球磨罐中以1800rpm的转速球磨3小时,取出产物,分别用去离子水洗3遍、乙醇洗3遍,离心分离得到的产物,放入干燥箱中80℃干燥4小时,即得前驱体。将所得前驱体和0.346克碳酸钾在研钵中混合均匀,放入通有h2/n2(体积:5%/95%)混合气体的管式炉中,550℃保温1小时,冷却即得k0.2wo3。
将所得k0.2wo3与0.397克碳酸钠在研钵中混合均匀,放入通有h2/n2(体积:5%/95%)混合气体的管式炉中,550℃保温1小时,冷却,即可得k0.2na0.3wo3样品。
实施例3:异质结构光催化剂k0.2li0.4wo3样品3的制备
称取7.098克仲钨酸铵和1.201克柠檬酸,放入行星球磨机的球磨罐中以1800rpm的转速球磨3小时,取出产物,分别用去离子水洗3遍、乙醇洗3遍,离心分离得到的产物,放入干燥箱中80℃干燥4小时,即得前驱体。将所得前驱体和0.346克碳酸钾在研钵中混合均匀,放入通有h2/n2(体积:5%/95%)混合气体的管式炉中,550℃保温1小时,冷却即得k0.2wo3。
将所得k0.2wo3与0.369克碳酸锂在研钵中混合均匀,放入通有h2/n2(体积:5%/95%)混合气体的管式炉中,550℃保温1小时,冷却,即可得k0.2li0.4wo3样品。
实施例4:异质结构光催化剂k0.2li0.4wo3样品4的制备
称取7.098克仲钨酸铵和1.201克柠檬酸,放入行星球磨机的球磨罐中以1800rpm的转速球磨3小时,取出产物,分别用去离子水洗3遍、乙醇洗3遍,离心分离得到的产物,放入干燥箱中80℃干燥4小时,即得前驱体。将所得前驱体和0.346克碳酸钾在研钵中混合均匀,放入通有h2/n2(体积:5%/95%)混合气体的管式炉中,550℃保温1小时,冷却,即得k0.2wo3。
将所得k0.2wo3与0.530克碳酸钠在研钵中混合均匀,放入通有h2/n2(体积:5%/95%)混合气体的管式炉中,550℃保温1小时,冷却,即可得k0.2na0.4wo3样品。
实施例5:异质结构光催化剂k0.2li0.4wo3样品5的制备
称取3.098克偏钨酸铵和1.201克柠檬酸,放入搅拌磨中以1400rpm的转速球磨4小时,取出产物,分别用去离子水洗3遍、乙醇洗3遍,离心分离得到的产物,放入干燥箱中80℃干燥4小时,即得前驱体。将所得前驱体和0.346克碳酸钾在研钵中混合均匀,放入通有氢气气体的管式炉中,500℃保温6小时,冷却,即得k0.2wo3。
将所得k0.2wo3与0.530克碳酸钠在研钵中混合均匀,放入通有氢气气体的管式炉中,500℃保温6小时,冷却,即可得k0.2na0.4wo3样品。
实施例6:异质结构光催化剂k0.2li0.4wo3样品6的制备
称取7.098克仲钨酸铵和0.938克酒石酸,放入砂磨机中以2500rpm的转速球磨6小时,取出产物,分别用去离子水洗3遍、乙醇洗3遍,离心分离得到的产物,放入干燥箱中80℃干燥4小时,即得前驱体。将所得前驱体和0.346克碳酸钾在研钵中混合均匀,放入通有一氧化碳气体的管式炉中,800℃保温0.5小时,冷却,即得k0.2wo3。
将所得k0.2wo3与0.530克碳酸钠在研钵中混合均匀,放入通有一氧化碳气体的管式炉中,800℃保温0.5小时,冷却,即可得k0.2na0.4wo3样品。
对比例:k0.2wo3样品的制备
称取8.246克二水合钨酸钠和1.576克二水合草酸,放入行星球磨机的球磨罐中以1800rpm的转速球磨3小时,取出产物,分别用去离子水洗3遍、乙醇洗3遍,离心分离得到的产物,放入干燥箱中80℃干燥4小时,即得前驱体。将所得前驱体和0.346克碳酸钾在研钵中混合均匀,放入通有h2/n2(体积:体积:5%/95%)混合气体的管式炉中,550℃保温1小时,冷却,即得k0.2wo3样品。
实验1:实施例1、2和对比例样品的x射线衍射/电镜扫描/电镜透射分析
上述实施例1、实施例2和对比例样品的x射线衍射图谱见图1。由图1可知,对比例样品的x射线特征衍射峰与k0.2wo3(jcpdsno.83-1331)完全吻合,可以判断为k0.2wo3;实施例1样品x射线特征衍射峰对应两种相分别为k0.2wo3(jcpdsno.83-1331)和li2w2o7(jcpdsno.73-0171);实施例2样品x射线特征衍射峰对应两种相分别为k0.2wo3(jcpdsno.83-1331)和na2w2o7(jcpdsno.70-0860)。
上述实施例1、实施例2和对比例样品的扫描电镜照片见图2。,其中图(a)为对比例样品,图(b)为实施例1样品,图(c)为实施例2样品。由图2可知,对比例样品主要由小颗粒团聚组成,实施例1样品有由颗粒向棒状生长的趋势,实施例2样品由于钠离子的掺入,形貌由小颗粒团聚体变成了棒状结构,形成的棒有长有短,形貌差异较大。
上述实施例1样品的透射电镜(a)和相应位置的高分辨率透射电镜(b)照片见图3。由图3可以看出,实施例1样品主要含有两种晶格条纹,(b)图中右上方局部放大图中的晶格间距为0.612nm,对应li2w2o7的(-110)晶面;而(b)图中右下方局部放大图中的晶格间距为0.320nm,对应k0.2wo3的(200)晶面,间接证明实施例1样品具有两个物相,并形成异质结构。
上述实施例2样品的透射电镜(a)和高分辨率透射电镜(b)照片见图4。由图4可以看出,实施例2样品主要为棒状结构,具有两种不同的晶格条纹间距,(b)图中晶格间距为0.320nm的晶格条纹对应k0.2wo3的(200)晶面;而(b)图中晶格间距为0.384nm所对应的是na2w2o7的(113)晶面,间接证明实施例2样品同样具有两个物相,并形成异质结构。
实验2:实施例1、2和对比例样品的光催化实验:
100mg实施例2和实施例1样品分别放入50ml内含50mg/l罗丹明b溶液中,恒定30℃,分别在紫外光、可见光和近红外光辐照下进行催化降解,同时以对比例所制备的样品在相同条件下进行对比试验,试验结果如下表;
由上表可知,实施例2样品和实施例1样品异质结光催化剂在紫外光、可见光和近红外光辐照下对溶液中的罗丹明b均显示强烈的去除性能,在同样的实验条件下,与对比例样品相比,其去除效率大幅度提高。
30℃时,实施例1、实施例2和对比例样品分别在紫外光(a)、可见光(b)和近红外光(c)辐照下去除溶液中rhb的变化曲线及其一级动力学速率常数柱形图见图5。
由图5可知,不管在紫外、可见还是近红外光波段下,锂离子或钠离子的掺入都提高了催化剂的催化活性。对比例样品在紫外、可见光和近红外光照射下的速率分别为0.0053min-1、0.00027min-1和0.00019min-1,而实施例1样品在紫外光(0.043min-1)、可见光(0.022min-1)和近红外光(0.021min-1)照射下rhb降解速率常数分别是对比例样品的8.11倍、81.5倍和110.5倍,实施例2样品在紫外-可见-近红外光照射120min后,相对应的rhb降解速率为0.02562min-1,0.01959min-1和0.01818min-1。
实验3:实施例1样品的光催化活性温度影响实验:
在19℃、30℃和40℃下,100mg实施例1样品放入50ml50mg/l罗丹明b溶液中,分别在紫外光、可见光和近红外光辐照下进行催化降解,三种光源照射时间均为180min。其去除溶液中rhb的变化曲线及其一级动力学速率常数柱形图见图6。
由图6可知,当温度较低时,实施例1样品光催化降解rhb的活性大大降低。在19℃下近红外光照射180min实施例1样品只去除了13.4%的rhb,去除率十分低,而40℃下120min时rhb的去除率就达到93%。在温度为19℃下,实施例1样品在可见光进行催化降解180min,rhb也只去除了49.7%,而在紫外光照射下rhb的去除率较高,与其他温度差不多,只是降解速度较慢。40℃时rhb的去除率与30℃时的相同,但降解速度相对应30℃快,降解时间短,这一性能在可见和紫外区域表现的更加明显,温度为40℃下30min后rhb的去除率达到最大值分别为94%和95.1%。不管在什么波段的太阳光下,随温度的增加实施例1的光催化速率也随之增加。在19℃下,其在紫外、可见和近红外照射下降解rhb的速率常数分别为0.01723min-1、0.00300min-1和0.00077min-1。而在40℃并在紫外光(0.05014min-1)、可见光(0.0441min-1)和近红外光(0.03406min-1)照射下的rhb降解速率常数分别是19℃下的2.9倍、14.7倍和44.2倍,以上数据显示实施例1样品分别在紫外光(a)、可见光(b)和近红外光(c)辐照下去除溶液中rhb呈现明显的光热协同增强效应。
实验4:实施例2样品的光催化活性温度影响实验:
在19℃、30℃和40℃下,100mg实施例2样品放入50ml50mg/l罗丹明b溶液中,分别在紫外光、可见光和近红外光辐照下进行催化降解,三种光源照射时间均为120min。其去除溶液中rhb的变化曲线及其一级动力学速率常数柱形图见图7。
由图7可知,在近红外和可见光下,温度为19℃时实施例2样品催化活性较低,rhb分别去除了28.6%和31.8%,在紫外光照射120min去除rhb的量略有减小为87.4%。随着温度的增加,催化剂的吸附量也随之增加,尤其是溶液温度由30℃变为40℃时,催化剂吸附溶液中rhb的量由21.5%变为84.6%,这说明温度的改变对实施例2样品吸附rhb的影响十分大。紫外和可见光区域的光催化速率大于近红外光区域,在紫外光辐照下19℃、30℃和40℃的降解速率分别为0.0167min-1、0.02562min-1和0.01807min-1,以上数据显示实施例2样品分别在紫外光(a)、可见光(b)和近红外光(c)辐照下去除溶液中rhb呈现明显的光热协同增强效应。
同样地,将实施例3-6所得到的样品3-6,进行x射线衍射/电镜扫描/电镜透射分析、并按照实验2、3所述步骤进行光催化实验和光催化活性温度影响实验,所得到的实验结果与实验1-3所述的实验结果基本一致。即:不管在紫外、可见还是近红外光波段下,因有锂离子的掺入提高了催化剂的催化活性;各样品分别在紫外光、可见光和近红外光辐照下,去除溶液中rhb均能呈现明显的光热协同增强效应。
以上对本发明的具体实施例进行了详细描述,但其只是作为范例,本发明并不限制于以上描述具体实施例。对于本领域技术人员而言,任何对本发明进行的等同修改和替代也都在本发明的范畴之中。因此,在不脱离本发明的精神和范围下所作的均等变换和修改,都涵盖在本发明范围内。
- 还没有人留言评论。精彩留言会获得点赞!