降低烟气中SO的制作方法
降低烟气中so
x
与no
x
的催化剂及其制备方法和应用以及烟气脱so
x
和no
x
的方法
技术领域
1.本发明涉及催化裂化技术领域,具体涉及能够同时降低烟气中so
x
与no
x
的催化剂及其制备方法和应用以及烟气同时脱so
x
和no
x
的方法。
背景技术:
2.催化剂表面结焦是导致催化裂化催化剂失活的主要原因,因此需要将催化剂高温氧化再生,烧掉表面的焦炭。在烧焦过程中,会产生so
x
和no
x
等,这些物质排放到大气中会造成大气污染。目前世界各国对so
x
和no
x
的排放限制越来越严格。
3.目前降低催化裂化再生烟气污染物排放的主要技术措施包括:再生器优化,使用助剂和烟气后处理,其中添加助剂的方法因具有操作灵活和无需投资设施费用等优点而得到普遍应用。目前脱硫脱硝助剂主要是单独脱除一种烟气污染物。例如:cn1334316a公开了一种含有镁铝尖晶石的组合物以及铈/钒的氧化物的硫转移剂,用于催化裂化烟气中脱除so
x
。cn101311248b提供了一种能够降低催化裂化再生烟气中no
x
排放的组合物,用于降低催化裂化烟气中no
x
。
4.上述专利文献在单独脱除再生烟气中so
x
和no
x
时,具有较好的脱除效果,但不能同时脱除氮氧化物与硫氧化物。为了同时满足两种污染物排放达标往往需要加大加注量,这也导致了助剂的添加总量过大,成本过高。
技术实现要素:
5.本发明的目的是为了克服现有技术无法同时脱除再生烟气中so
x
和no
x
的缺陷,提供一种能够同时降低烟气中so
x
与no
x
的催化剂及其制备方法和应用以及烟气同时脱so
x
和no
x
的方法。本发明提供的催化剂能够同时脱除so
x
和no
x
,操作简单,且催化剂制备方法简单。
6.为了实现上述目的,本发明第一方面提供一种能够同时降低烟气中so
x
与no
x
的催化剂,该催化剂包括载体和负载在载体上的选自稀土族金属的第一活性组分、选自vb、viii、ib、iib族非贵金属的第二活性组分、选自iia族金属的第三活性组分以及贵金属组分;以催化剂总重量为基准,所述载体的含量为25-93重量%,以氧化物计,所述第一活性组分的含量为4-60重量%,所述第二活性组分的含量为2-30重量%,所述第三活性组分的含量为1-30重量%,以元素计,所述贵金属组分的含量为0.01-2重量%。
7.优选地,以金属元素计,所述第一活性组分与所述第二活性组分的摩尔比为(0.4-12):1,优选为(0.5-8):1,进一步优选为(1-4):1。
8.本发明第二方面提供一种制备能够同时降低烟气中so
x
与no
x
的催化剂的方法,该方法包括如下步骤:
9.(1)提供含有选自稀土族金属的第一活性组分前驱体、选自vb、viii、ib、iib族非贵金属的第二活性组分前驱体以及选自iia族金属的第三活性组分前驱体的前驱体溶液;
10.(2)将所述前驱体溶液与共沉淀剂进行共沉淀反应,然后进行干燥和焙烧;
11.(3)将步骤(2)得到的固体产物与载体和/或载体的前驱体以及任选地贵金属组分前驱体混合打浆,得到浆液,将浆液进行干燥和焙烧;
12.该方法还任选地包括:(4)以含有贵金属组分前驱体的溶液作为浸渍液,对步骤(3)得到的固体产物进行浸渍,然后进行干燥和/或焙烧;
13.第一活性组分前驱体、第二活性组分前驱体、第三活性组分前驱体、载体和/或载体的前驱体以及贵金属组分前驱体的用量使得制得的催化剂,以催化剂总重量为基准,所述载体的含量为25-93重量%,以氧化物计,所述第一活性组分的含量为4-60重量%,所述第二活性组分的含量为2-30重量%,所述第三活性组分的含量为1-30重量%,以元素计,所述贵金属组分的含量为0.01-2重量%。
14.本发明第三方面提供上述方法制备得到的能够同时降低烟气中so
x
与no
x
的催化剂。
15.本发明第四方面提供上述能够同时降低烟气中so
x
与no
x
的催化剂在含有脱so
x
和no
x
的烟气同时脱so
x
和no
x
反应中的应用。
16.本发明第五方面提供一种烟气同时脱so
x
和no
x
的方法,该方法包括:
17.将烟气与上述的能够同时降低烟气中so
x
与no
x
的催化剂接触。
18.本发明提供的催化剂将特定量的活性组分配合使用,能够同时降低烟气中so
x
和no
x
的排放。如实施例1中,在0.5h内本发明提供的用于同时降低烟气中so
x
与no
x
的催化剂,在混合进no和so2时,no污染物脱除率达到65%,so2污染物的脱除率达到70%,与现有技术制备的催化剂相在相同的评价条件下比较,在混合进no和so2的反应中的污染物脱除率,明显优于单独进so2气和单独进no气的反应。
具体实施方式
19.在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
20.在本发明中,所述能够同时降低烟气中so
x
与no
x
的催化剂指的是该催化剂能够用于同时脱除降低烟气中so
x
与no
x
,降低烟气中so
x
与no
x
的含量。
21.本发明第一方面提供一种能够同时降低烟气中so
x
与no
x
的催化剂,该催化剂包括载体和负载在载体上的选自稀土族金属的第一活性组分、选自vb、viii、ib、iib族非贵金属的第二活性组分、选自iia族金属的第三活性组分以及贵金属组分;以催化剂总重量为基准,所述载体的含量为25-93重量%,以氧化物计,所述第一活性组分的含量为4-60重量%,所述第二活性组分的含量为2-30重量%,所述第三活性组分的含量为1-30重量%,以元素计,所述贵金属组分的含量为0.01-2重量%。
22.根据本发明,常规定义的稀土族金属组分均可用于本发明,优选所述第一活性组分选自镧、铈、镨和钕中的一种或几种,更优选为镧和/或铈,最优选为镧。采用该种方式能够进一步提高催化剂的脱so
x
与no
x
的性能。
23.所述vb族非贵金属组分可以选自钒、铌和钽中的至少一种;所述viii族非贵金属
组分可以选自铁、钴和镍中的至少一种;所述ib族非贵金属组分可以为铜;所述iib族非贵金属组分可以选自锌、镉和汞中的至少一种。
24.优选情况下,所述第二活性组分选自铁、钴、镍、铜、锌和钒中的一种或几种,更优选为钴和/或铁,最优选为钴。
25.在本发明中,所述第三活性组分可以选自铍、镁、钙、锶和钡中的一种或几种,更优选为镁。
26.所述贵金属组分可以选自钌、铑、铼、铂、钯、银、铱和金中的一种或几种,优选为铂、钯和铑中的一种或几种,最优选为钯。本发明的发明人在研究过程中发现,将钯与其他活性组分配合使用,更有利于脱除烟气中的so
x
与no
x
。
27.在本发明提供的催化剂中,所述载体选择范围较宽,优选情况下,所述载体选自氧化铝、氧化硅-氧化铝、沸石、尖晶石、高岭土、硅藻土、珍珠岩和钙钛矿中的至少一种,进一步优选为氧化铝。
28.对所述氧化铝的晶型没有特别的限制,包括但不限于γ-氧化铝、δ-氧化铝、η-氧化铝、ρ-氧化铝、κ-氧化铝和χ-氧化铝。
29.根据本发明的一种优选实施方式,以催化剂总重量为基准,所述载体的含量为40-87重量%,以氧化物计,所述第一活性组分的含量为8-50重量%,所述第二活性组分的含量为3-20重量%,所述第三活性组分的含量为1-20重量%,以元素计,所述贵金属组分的含量为0.02-1.5重量%;
30.优选地,以催化剂总重量为基准,所述载体的含量为45-80重量%,以氧化物计,所述第一活性组分的含量为8-40重量%,所述第二活性组分的含量为3-15重量%,所述第三活性组分的含量为2-15重量%,以元素计,所述贵金属组分的含量为0.03-1.2重量%。
31.根据本发明,优选地,以金属元素计,所述第一活性组分与所述第二活性组分的摩尔比为(0.4-12):1,优选为(0.5-8):1,进一步优选为(1-4):1。本发明的发明人在研究过程中发现,将二者在特别比例下复配,能够起到更好的协同效果,更有利于脱除烟气中的so
x
与no
x
。
32.催化剂中组分含量均采用x射线荧光光谱(xrf)法测定,具体参见石油化工分析方法(ripp实验方法),杨翠定等编,科学出版社1990年出版。
33.本发明对上述催化剂的制备方法选择范围较宽,只要能够得到上述催化剂即可,可以利用共沉淀法,也可以为溶胶凝胶法。
34.本发明第二方面提供一种制备能够同时降低烟气中so
x
与no
x
的催化剂的方法,该方法包括如下步骤:
35.(1)提供含有选自稀土族金属的第一活性组分前驱体、选自vb、viii、ib、iib族非贵金属的第二活性组分前驱体以及选自iia族金属的第三活性组分前驱体的前驱体溶液;
36.(2)将所述前驱体溶液与共沉淀剂进行共沉淀反应,然后进行干燥和焙烧;
37.(3)将步骤(2)得到的固体产物与载体和/或载体的前驱体以及任选地贵金属组分前驱体混合打浆,得到浆液,将浆液进行干燥和焙烧;
38.该方法还任选地包括:(4)以含有贵金属组分前驱体的溶液作为浸渍液,对步骤(3)得到的固体产物进行浸渍,然后进行干燥和/或焙烧;
39.第一活性组分前驱体、第二活性组分前驱体、第三活性组分前驱体、载体和/或载
体的前驱体以及贵金属组分前驱体的用量使得制得的催化剂,以催化剂总重量为基准,所述载体的含量为25-93重量%,以氧化物计,所述第一活性组分的含量为4-60重量%,所述第二活性组分的含量为2-30重量%,所述第三活性组分的含量为1-30重量%,以元素计,所述贵金属组分的含量为0.01-2重量%。
40.根据本发明提供的方法,优选地,第一活性组分前驱体、第二活性组分前驱体、第三活性组分前驱体、载体和/或载体的前驱体以及贵金属组分前驱体的用量使得制得的催化剂,以催化剂总重量为基准,所述载体的含量为40-87重量%,以氧化物计,所述第一活性组分的含量为8-50重量%,所述第二活性组分的含量为3-20重量%,所述第三活性组分的含量为1-20重量%,以元素计,所述贵金属组分的含量为0.02-1.5重量%;
41.优选地,第一活性组分前驱体、第二活性组分前驱体、第三活性组分前驱体、载体和/或载体的前驱体以及贵金属组分前驱体的用量使得制得的催化剂,以催化剂总重量为基准,所述载体的含量为45-80重量%,以氧化物计,所述第一活性组分的含量为8-40重量%,所述第二活性组分的含量为3-15重量%,所述第三活性组分的含量为2-15重量%,以元素计,所述贵金属组分的含量为0.03-1.2重量%。
42.根据本发明提供的方法,第一活性组分、第二活性组分、第三活性组分以及所述贵金属组分和载体的具体种类的选择范围如上文第一方面所述,在此不再赘述。
43.根据本发明的一种优选实施方式,以金属元素计,第一活性组分前驱体与第二活性组分前驱体的摩尔比为(0.4-12):1,优选为(0.5-8):1,进一步优选为(1-4):1。
44.本发明对步骤(1)提供所述前驱体溶液的方法没有特别的限定,只要使得各金属组分前驱体混合均匀即可。例如可以将各金属组分前驱体溶于水中,充分搅拌均匀。
45.根据本发明,优选地,第一活性组分前驱体、第二活性组分前驱体以及第三活性组分前驱体可以各自独立地选自各金属组分的水溶性盐,如硝酸盐、氯化物、氯酸盐或硫酸盐等,优选为硝酸盐和/或氯化物。
46.根据本发明,优选地,所述贵金属组分前驱体选自硝酸钯、氯化钯、氯铂酸和氯化铑中的至少一种,优选为硝酸钯和/或氯化钯。
47.本发明对所述共沉淀剂的种类和用量没有特别的限定,只要能够使得共沉淀反应顺利进行即可。所述共沉淀剂的种类可以为本领域的常规选择,优选地,所述共沉淀剂为碳酸盐,进一步优选选自碳酸铵、碳酸钾和碳酸钠中的至少一种,更优选为碳酸铵。
48.步骤(2)中,共沉淀剂可以以溶液的形式引入,与所述前驱体溶液进行共沉淀反应。本发明对所述前驱体溶液和共沉淀剂的溶液的浓度没有特别的限定,只要溶液浓度小于在制备时的溶解度,从而确保能够充分发生所述共沉淀反应即可。
49.优选地,所述共沉淀反应在ph为8-10,优选8.5-9.5条件下进行。所述共沉淀反应的ph可以通入加入酸和/或碱进行调节,对其具体种类没有特别的限定,例如可以为氨水。
50.根据本发明,还包括将共沉淀反应得到的反应产物进行固液分离(例如可以为过滤或者是离心分离),以得到固体产物,然后进行所述干燥和焙烧。
51.本发明中所述贵金属组分可以在步骤(3)中引入,也可以在步骤(4)中引入,还可以部分在步骤(3)引入,部分在步骤(4)引入,优选通过在步骤(4)中引入,该种优选实施方式更有利于贵金属的分散。
52.所述载体前驱体可以为任何能够通过后续焙烧转化为载体的物质,本领域技术人
员可以通过载体的具体种类进行适当选择,本发明在此不再赘述。例如,氧化铝的前驱体可以选自铝的各种溶胶或凝胶,或者氢氧化铝。所述氢氧化铝可以选自三水铝石、湃铝石、诺水铝石、硬水铝石、薄水铝石和拟薄水铝石中的至少一种。
53.根据本发明提供的方法,所述载体为氧化铝,优选地,在打浆之前,对载体和/或载体的前驱体进行酸化处理,所述酸化处理可以按照本领域常规技术手段进行,进一步优地,所述酸化处理使用的酸为盐酸。
54.本发明对所述酸化处理的条件的选择范围较宽,优选地,所述酸化处理的条件包括:酸铝比为0.12-0.22:1,时间为20-40min。
55.在本发明中,无特殊说明情况下,所述酸铝比是指以36重量%的浓盐酸计的盐酸与以干基计的氧化铝的前驱体的质量比。
56.所述酸化胶溶处理的具体实施方式可以为:将氧化铝前驱体加入水中打浆分散。
57.本发明提供的方法,对所述步骤(2)得到的固体产物、载体和/或载体的前驱体以及任选地贵金属组分前驱体混合打浆的方法没有特别的限定,对上述物料的加入顺序同样没有限定,只要将上述物料以及水接触混合均匀即可。当打浆过程中还含有贵金属组分前驱体时,具体混合打浆过程可以包括:将贵金属组分前驱体(可以以溶液形式引入)加入酸化后的载体混合打浆,再将步骤(2)得到的固体产物加入,然后对浆液进行干燥和焙烧。
58.根据本发明,优选地,步骤(3)所述浆液的固含量为6-38重量%。
59.根据本发明提供的方法,步骤(3)所述干燥优选为喷雾干燥,在本发明中,所述喷雾干燥可以按照本领域常规技术手段进行,本发明对此没有特别的限定。本领域技术人员可以根据目标催化剂的平均粒径选择适当的喷雾干燥条件,优选喷雾干燥的条件使得喷雾干燥得到的颗粒的平均粒径为60-80μm,粒径分布范围主要在20-100μm。
60.根据本发明提供的方法,对步骤(4)中所述浸渍没有特别的限定,可以按照本领域常规技术手段进行,可以为饱和浸渍,也可以为过量浸渍,优选为过量浸渍。本领域技术人员可以根据目标产物中贵金属的含量选择适当的操作。
61.根据本发明,优选地,步骤(4)中,将贵金属组分前驱体在酸溶液中水解以提供所述浸渍液。具体地,还可以在所述水解以后,进行稀释(可以加水)或者提浓(可以进行蒸发),然后进行所述浸渍以提供特定贵金属组分负载量的催化剂。
62.优选地,所述酸选自可溶于水的无机酸和/或有机酸,优选选自盐酸、硝酸、磷酸和醋酸中至少一种。
63.根据本发明,优选地,所述酸的用量使得浸渍液的ph值小于6.0,优选为2-5。采用该种优选实施方式更有利于活性组元均匀分散及改善成品催化剂的耐磨损强度。
64.本发明可以通过对浸渍后得到的混合物进行过滤得到所述固体产物。所述过滤可以按照本领域常规技术手段进行。
65.本发明步骤(4)中可以仅对固体产物进行干燥,也可以仅对固体产物进行焙烧,还可以对所述固体产物进行干燥后进行焙烧,本发明对此没有特别的限定,优选对所述固体产物进行干燥后进行焙烧。本发明对所述干燥和焙烧的条件没有特别的限定,可以按照本领域常规技术手段进行。例如,干燥的条件可以包括:温度为60-200℃,时间为2-10h。
66.本发明提供方法中,不同步骤的焙烧条件相同或不同,优选地,步骤(2)、步骤(3)和步骤(4)所述的焙烧条件各自独立地包括:温度为300-800℃,时间为0.5-8h。所述焙烧可
以在空气或惰性气氛(例如氮气)中进行,本发明对此没有特别的限制,优选在空气气氛下进行。
67.本发明第三方面提供上述方法制备得到的能够同时降低烟气中so
x
与no
x
的催化剂。
68.本发明提供的催化剂特别适合于含有so
x
和no
x
的烟气的处理。基于此,本发明第四方面提供上述催化剂在含有脱so
x
和no
x
的烟气同时脱so
x
和no
x
反应中的应用。
69.本发明第五方面提供一种烟气同时脱so
x
与no
x
的方法,该方法包括:
70.将烟气与上述的能够同时降低烟气中so
x
与no
x
的催化剂接触。
71.根据本发明的一种优选实施方式,所述接触的条件包括:温度为500-800℃,压力为0.01-4mpa,催化裂化再生烟气的体积空速为200-20000h-1
;进一步优选地,温度为550-750℃,压力为0.02-0.1mpa,催化裂化再生烟气的体积空速为500-10000h-1
。在本发明中,没有特殊限定的情况下,所述压力为表压。
72.优选地,所述烟气中,so
x
的含量为0.001-0.5体积%,no
x
的含量为0.001-0.3体积%。
73.优选地,所述烟气中,so
x
与no
x
的体积含量比值为(1-1.4):1,优选为(1-1.2):1。采用该种优选实施方式更有利于提高so
x
与no
x
的脱除效率。
74.本发明对所述烟气的选择范围较宽,本发明提供的方法适用于任何同时含有so
x
和no
x
的烟气。
75.优选地,所述烟气为催化裂化再生烟气。所述催化裂化再生烟气中除了含有so
x
和no
x
之外,还可以含有co、co2、h2o组分。
76.在本发明中,无特殊说明情况下,所述ppm指的是体积浓度。
77.以下将通过实施例对本发明进行详细描述。
78.以下实施例中,催化剂中组分含量均采用x射线荧光光谱(xrf)法测定,具体参见石油化工分析方法(ripp实验方法),杨翠定等编,科学出版社1990年出版。对比例和实施例中所用原料:硝酸镧(分析纯,aladdin biochemical公司)、硝酸镁(分析纯,国药集团化学试剂有限公司)、硝酸钴(分析纯,北京伊诺凯科技有限公司)、碳酸铵(分析纯,北京化工厂)、氨水(分析纯,25%,天津市大茂化学厂)、氯化钯(中国医药公司北京采购供应站),盐酸(北京化工厂),ox50-sio2(中石化催化剂公司)。
79.实施例1
80.在烧杯中称取390ml去离子水,搅拌下加入以la2o3质量计为30g的硝酸镧、以mgo质量计为4g的硝酸镁、以co2o3质量计为5.5g的硝酸钴直至完全溶解。称取碳酸铵59.25g溶于250ml去离子水中,搅拌至充分溶解,将金属硝酸盐混合溶液在搅拌状态下加入到碳酸铵溶液中,并加入一定量氨水维持溶液ph值在9。将沉淀完全的混合物进行抽滤,并用去离子水淋洗,将抽滤得到的滤饼混合物在120℃下烘干,在空气气氛700℃焙烧6小时后,研磨得到活性金属前驱体。
81.称取以al2o3质量计为40g的铝石,加入380ml水及6g的36重量%的浓盐酸,进行打浆。称取20g的活性金属前驱体加入到酸化后的无机氧化物基质中混合搅拌,将浆液在200℃下进行干燥,并且在空气气氛700℃下焙烧4小时,得到催化剂微球半成品。非贵金属活性组分占所制备微球催化剂半成品的质量百分比为33%。
82.称取钯的前驱体与稀盐酸按质量比1:1互溶,加去离子水稀释,配置成浓度为5.6g/l的氯化钯溶液,ph为2,称取15g的催化剂微球半成品,按含钯质量为0.0045g称取一定量浓度为5.6g/l的氯化钯溶液。将氯化钯溶液作为浸渍液浸渍到上述催化剂微球半成品,得到固体产物,然后对固体产物在120℃下进行干燥,然后在空气气氛700℃下焙烧4小时,即得到催化剂s-1。
83.实施例2
84.在烧杯中称取220ml去离子水,搅拌下加入以la2o3质量计为10g的硝酸镧、以mgo质量计为7g的硝酸镁、以co2o3质量计为5g的硝酸钴和直至完全溶解。称取碳酸铵33g溶于150ml去离子水中,搅拌至充分溶解,将金属硝酸盐混合溶液在搅拌状态下加入到碳酸铵溶液中,并加入一定量氨水维持溶液ph值在9。将沉淀完全的混合物进行抽滤,并用去离子水淋洗,将抽滤得到的滤饼混合物在120℃下烘干,在空气气氛700℃焙烧6小时后,研磨得到活性金属前驱体。
85.称取以al2o3质量计为40g的铝石,加入330ml水及6g的36重量%的浓盐酸,进行打浆。称取10g的活性金属前驱体加入到酸化后的无机氧化物基质中混合搅拌,将浆液在200℃下进行干燥,并且在空气气氛700℃下焙烧4小时,得到催化剂微球半成品。非贵金属活性组分占所制备微球催化剂半成品的质量百分比为20%。
86.称取钯的前驱体与稀盐酸按质量比1:1互溶,加去离子水稀释,配置成浓度为5.6g/l的氯化钯溶液,ph为2,称取15g的催化剂微球半成品,按含钯质量为0.0030g称取一定量浓度为5.6g/l的氯化钯溶液。将氯化钯溶液作为浸渍液浸渍到上述催化剂微球半成品,得到固体产物,然后对固体产物在120℃下进行干燥,然后在空气气氛700℃下焙烧4小时,即得到的催化剂记为s-2。
87.实施例3
88.在烧杯中称取390ml去离子水,搅拌下加入以la2o3质量计为30g的硝酸镧、以mgo质量计为5g的硝酸镁、以co2o3质量计为4g的硝酸钴直至完全溶解。称取碳酸铵58.5g溶于250ml去离子水中,搅拌至充分溶解,将金属硝酸盐混合溶液在搅拌状态下加入到碳酸铵溶液中,并加入一定量氨水维持溶液ph值在9。将沉淀完全的混合物进行抽滤,并用去离子水淋洗,将抽滤得到的滤饼混合物在120℃下烘干,在空气气氛700℃焙烧6小时后,研磨得到活性金属前驱体。
89.称取以al2o3质量计为40g的铝石,加入380ml水及6g的36重量%的浓盐酸,进行打浆。称取20g的活性金属前驱体加入到酸化后的无机氧化物基质中混合搅拌,将浆液在200℃下进行干燥,并且在空气气氛700℃下焙烧4小时,得到催化剂微球半成品。非贵金属活性组分占所制备微球催化剂半成品的质量百分比为33%。
90.称取钯的前驱体与稀盐酸按质量比1:1互溶,加去离子水稀释,配置成浓度为5.6g/l的氯化钯溶液,ph为2,称取15g的催化剂微球半成品,按含钯质量为0.0030g称取一定量浓度为5.6g/l的氯化钯溶液。将氯化钯溶液作为浸渍液浸渍到上述催化剂微球半成品,得到固体产物,然后对固体产物在120℃下进行干燥,然后在空气气氛700℃下焙烧4小时,即得到的催化剂记为s-3。
91.实施例4
92.按照实施例1的方法,不同的是,减少铝石的用量,将非贵金属活性组分占所制备
微球催化剂半成品的质量百分比调整为50%,即称取以al2o3质量计为20g的铝石和20g的活性金属前驱体,得到催化剂s-4。
93.实施例5
94.在烧杯中称取390ml去离子水,搅拌下加入以la2o3质量计为34g的硝酸镧、以mgo质量计为4g的硝酸镁、以co2o3质量计为1g的硝酸钴直至完全溶解。称取碳酸铵58.5g溶于250ml去离子水中,搅拌至充分溶解,将金属硝酸盐混合溶液在搅拌状态下加入到碳酸铵溶液中,并加入一定量氨水维持溶液ph值在9。将沉淀完全的混合物进行抽滤,并用去离子水淋洗,将抽滤得到的滤饼混合物在120℃下烘干,在空气气氛700℃焙烧6小时后,研磨得到活性金属前驱体。
95.称取以al2o3质量计为40g的铝石,加入380ml水及6g的36重量%的浓盐酸,进行打浆。称取20g的活性金属前驱体加入到酸化后的无机氧化物基质中混合搅拌,将浆液在200℃下进行干燥,并且在空气气氛700℃下焙烧4小时,得到催化剂微球半成品。非贵金属活性组分占所制备微球催化剂半成品的质量百分比为33%。
96.称取钯的前驱体与稀盐酸按质量比1:1互溶,加去离子水稀释,配置成浓度为5.6g/l的氯化钯溶液,ph为2,称取15g的催化剂微球半成品,按含钯质量为0.0030g称取一定量浓度为5.6g/l的氯化钯溶液。将氯化钯溶液作为浸渍液浸渍到上述催化剂微球半成品,得到固体产物,然后对固体产物在120℃下进行干燥,然后在空气气氛700℃下焙烧4小时,即得到催化剂s-5。
97.实施例6
98.在烧杯中称取480ml去离子水,搅拌下加入以la2o3质量计为22g的硝酸镧、以mgo质量计为4g的硝酸镁、以co2o3质量计为22g的硝酸钴直至完全溶解。称取碳酸铵72g溶于300ml去离子水中,搅拌至充分溶解,将金属硝酸盐混合溶液在搅拌状态下加入到碳酸铵溶液中,并加入一定量氨水维持溶液ph值在9。将沉淀完全的混合物进行抽滤,并用去离子水淋洗,将抽滤得到的滤饼混合物在120℃下烘干,在空气气氛700℃焙烧6小时后,研磨得到活性金属前驱体。
99.称取以al2o3质量计为40g的铝石,加入380ml水及6g的36重量%的浓盐酸,进行打浆。称取20g的活性金属前驱体加入到酸化后的无机氧化物基质中混合搅拌,将浆液在200℃下进行干燥,并且在空气气氛700℃下焙烧4小时,得到催化剂微球半成品。非贵金属活性组分占所制备微球催化剂半成品的质量百分比为33%。
100.称取钯的前驱体与稀盐酸按质量比1:1互溶,加去离子水稀释,配置成浓度为5.6g/l的氯化钯溶液,ph为2,称取15g的催化剂微球半成品,按含钯质量为0.0045g称取一定量浓度为5.6g/l的氯化钯溶液。将氯化钯溶液作为浸渍液浸渍到上述催化剂微球半成品,得到固体产物,然后对固体产物在120℃下进行干燥,然后在空气气氛700℃下焙烧4小时,即得到催化剂s-6。
101.实施例7
102.按照实施例1的方法,不同的是,将氯化钯溶液替换为等浓度的氯化钌溶液,且制得的催化剂中,贵金属含量不变,即得到催化剂s-7。
103.实施例8
104.按照实施例1的方法,不同的是,以氧化物计,将硝酸镧替换为等质量的硝酸铈;以
氧化物计,将硝酸钴替换为等质量的硝酸铁,即得到催化剂s-8。
105.对比例1
106.称取以la2o3质量计为30g的硝酸镧溶解于烧杯中,称取45g的碳酸铵完全溶解于烧杯中,在搅拌条件下,将硝酸镧溶液在搅拌状态下加入到碳酸铵溶液中,并加入一定量氨水维持溶液ph值在9。将上述得到的混合物进行抽滤,将抽滤得到的滤饼混合物在120℃直至烘干,在空气气氛700℃焙烧6小时后,研磨得到催化前驱体l。
107.称取以co2o3质量计为5g的硝酸钴完全溶解于烧杯中,称取7.5g的碳酸铵完全溶解于烧杯中,在搅拌条件下,将硝酸钴溶液在搅拌状态下加入到碳酸铵溶液中,并加入一定量氨水维持溶液ph值在9。将上述得到的混合物进行抽滤,将抽滤得到的滤饼混合物在120℃直至烘干,在空气气氛700℃焙烧6小时后,研磨即制得活性金属前驱体c。
108.将前两步得到的活性金属前驱体l和活性金属前驱体c充分机械混合,得到混合前驱体。
109.称取以al2o3质量计为40g的铝石,加入380ml水及6g的36重量%的浓盐酸,进行打浆。取20g的混合前驱体加入到酸化后的无机氧化物基质中混合搅拌,将浆液在120℃下进行干燥,在空气气氛700℃下焙烧4小时,即得到催化剂d-1。
110.对比例2
111.催化剂按如下方法制备:分别称取15g ox50(sio2)粉末和按含钯质量为0.0045g称取一定量的实施例1中配置的氯化钯溶液。将氯化钯溶液加入到ox50粉末中,并通过不断搅拌使其混合均匀。将得到的混合物置于120℃条件下的烘箱内直至烘干,并且在空气气氛700℃下焙烧4小时,即得到催化剂d-2。
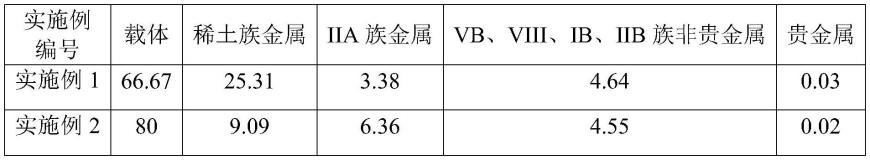
112.表1催化剂的组成(重量百分含量,%)
[0113][0114][0115]
本发明中催化剂活性评价标准以反应产物中so
x
和no
x
浓度变化作为衡量,产物中so
x
和no
x
含量采用ft-ir傅里叶红外烟气分析仪测量,采用固定床微反实验装置进行评价。催化剂活性评价结果以转化率表示。
[0116]
转化率计算方法:
[0117][0118]
其中,c
(inlet)
指的是实验装置入口so
x
或no
x
的浓度;c
(outlet)
指的是实验装置出口so
x
或no
x
的浓度。
[0119]
试验例1
[0120]
本试验例用于对上述实施例和对比例提供的催化剂在烟气中同时降低no和so2排放的作用进行评价。所述催化裂化反应-再生评价在小型固定床模拟烟气装置上进行,催化剂装填量为1.8g,反应温度为680℃,压力为0.03mpa,原料气体积流量(标况)为1200ml/min,体积空速约为10000h-1
。原料气含有1200ppm的no,1200ppm的so2,余量为n2。通过在线红外分析仪分析气体产物,得到反应后so2和no浓度。评价时间为0.5h的结果列于表2,评价时间为1.5h的结果列于表3。
[0121]
表20.5h内不同催化剂的性能比较
[0122]
0.5h总转化率(%)组合-no组合-so2单独-no单独-so2s-16570《260s-24450《247s-35360《257s-47973《263s-53862《261s-63151《250s-76167《257s-85669《262d-1《233《232d-210《210《2
[0123]
注:表2中单独-no和单独-so2分别指的是原料气中仅含有1200ppmno或者1200ppm so2[0124]
由表2可见,在前0.5h内,本发明提供的用于同时降低烟气中so
x
与no
x
的催化剂,与现有技术制备的催化剂相在相同的评价条件下比较,在混合进no和so2的反应中的污染物脱除率,明显优于单独进so2气和单独进no气的反应。说明本发明提供的催化剂具有较高的催化转化活性,尤其是在组合催化过程中no的催化转化活性得到了大幅提升。
[0125]
表3 1.5h内不同催化剂的性能比较
[0126][0127][0128]
注:表3中单独-no和单独-so2分别指的是原料气中仅含有1200ppmno或者1200ppm so2[0129]
由表3可见,在1.5h内,本发明提供的用于同时降低烟气中so
x
与no
x
的催化剂与现有技术制备的催化剂相在相同的评价条件下比较,在混合进no和so2的反应中的污染物脱除率,明显优于单独进so2气和单独进no气的反应。1.5h总转化率与0.5h总转化率相比有小幅度的下降,尤其在so2脱除转化率上,降幅非常小,说明iia族金属组分的引入能有效缓解硫致失活的问题,有效提升了催化剂的反应稳定性。
[0130]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1