合成丁二酸酐的金属碳化物催化剂及其制备方法和应用
1.本发明涉及有机合成技术领域,尤其涉及一种合成丁二酸酐的金属碳化物催化剂。
2.本发明还涉及所述催化剂的制备方法及应用。
背景技术:
3.白色污染已成为影响生态环境的重要污染物,开发和使用生物可降解塑料是解决这一问题的关键。在聚己内酯、聚乳酸、聚丁二酸丁二醇酯等各类可降解塑料产品中,聚丁二酸丁二醇酯具有耐热性能、机械性能和加工性能优越等优点,需求量逐年增加。
4.丁二酸酐是生产聚丁二酸丁二醇酯的重要原料之一,聚丁二酸丁二醇酯产业的快速发展必将拉动丁二酸酐市场需求。此外,丁二酸酐还广泛应用于医药、新材料、染料、食品工业、日用化学品工业等领域。在染料工业行业,丁二酸酐用于漆料、顺丁烯酸酯、聚酯和醇酸树脂等产品的生产;在纺织品工业行业,丁二酸酐可以用于生产染色材料;在表面活性剂行业,丁二酸酐可用于去污剂、破乳剂及肥皂的生产;在医药行业,丁二酸酐可以用于维生素、磺胺药、解毒剂、止血药、镇静剂等产品的生产;在食品行业,丁二酸酐可以明显提升食品的品质和加工质量;在农药行业,丁二酸酐可用于植物生长抑制剂和除草剂的生产。
5.丁二酸酐的工业生产方法包括电解合成-脱水法、生物发酵-脱水法和催化加氢法。电解合成-脱水法电耗高、电极腐蚀严重、脱水工艺能耗高、生产成本高。生物发酵法存在发酵分离过程复杂、脱水工艺成本高、废水量大等问题。加氢合成法可由顺酐一步加氢合成丁二酸酐,无需脱水过程,且产品纯度高、反应过程简捷高效。
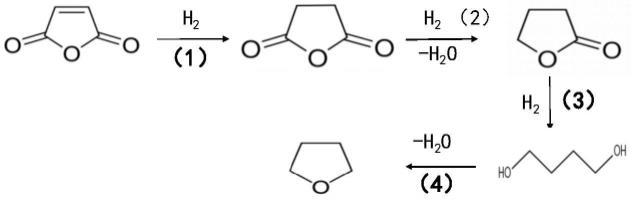
6.顺酐加氢生成丁二酸酐的反应路径为:顺酐加氢生成丁二酸酐(1)。过程的副反应包括丁二酸酐进一步加氢生成γ-丁内酯(2),γ-丁内酯进一步加氢则生成1,4-丁二醇(3),部分1,4-丁二醇进一步脱水生成四氢呋喃(4)。具体反应式如下:
[0007][0008]
可以看出,以上副反应发生的过程中,有副产物水的生成。副产物水对顺酐加氢反应有两个重要的影响。第一,原料顺酐和产物丁二酸酐在水存在的反应条件下会分别水解为顺丁烯二酸和丁二酸,会对催化剂造成腐蚀性,影响催化剂反应活性和使用寿命。第二,顺丁烯二酸极易被催化剂的酸性位催化转变为反丁烯二酸(富马酸),反应过程如下。富马酸溶解性差,其不断生成和累积很容易堵塞反应管道,影响反应的正常进行,甚至发生装置被迫停止运行。
[0009][0010]
要解决以上问题,要求催化剂具有以下性能:第一,催化剂要具有耐酸腐蚀性,能够在酸性反应介质下长期运行;第二,催化剂制备过程中要抑制载体上的酸性位,并使催化剂具备一定的碱性,从而抑制副产物富马酸的生成。
[0011]
目前的顺酐加氢催化剂通常以ni、co、mo等过渡金属为活性组分,如专利cn103769117a、cn104399469a、cn114289024a等。反应体系存在的顺丁烯二酸和丁二酸等有机酸对金属有一定的腐蚀性,而以上专利没有涉及催化剂耐腐蚀性的内容。
技术实现要素:
[0012]
本发明的目的在于提供一种合成丁二酸酐的金属碳化物催化剂,同时具有耐酸腐蚀性、加氢合成丁二酸酐性能和抑制富马酸生成的性能。
[0013]
为实现以上目的,本发明采用以下具体技术方案:
[0014]
一种合成丁二酸酐的金属碳化物催化剂,包括过渡金属碳化物、助剂金属氧化物和载体,其中,催化剂具有碱性位。
[0015]
所述催化剂上金属碳化物具有加氢活性位,同时在载体上引入碱性位抑制富马酸生成,并使两种活性位合理匹配。
[0016]
按照重量比,所述催化剂中,过渡金属碳化物占比为7.9-13.5wt%,助剂金属氧化物占比为0.3-1.2wt%,载体占比为86.2-91.0%;催化剂中碱性位浓度为0.25-0.53mmol/g。
[0017]
所述过渡金属碳化物为ni3c、co2c、moc的一种或几种;所述助剂为 ceo2和/或la2o3,所述载体为活性炭、氧化钛、二氧化硅或氧化锆。
[0018]
ni3c、co2c、moc等金属碳化物具有加氢功能;碱性位的作用是抑制副产物富马酸的生成;ceo2或la2o3可以促进金属碳化物和载体的协同作用,有效提高催化剂的加氢功能。
[0019]
本发明还提供一种所述合成丁二酸酐的金属碳化物催化剂的制备方法,包括如下步骤:
[0020]
s1:将助剂金属盐加入到去离子水中,然后浸渍于载体上,干燥得到样品一。
[0021]
进一步地,所述助剂金属盐与载体的质量比为(0.15-0.50):(12-14)。
[0022]
可选地,所述助剂金属盐为硝酸铈和硝酸镧中的一种;
[0023]
进一步地,所述干燥为多次干燥;优选地,所述多次干燥为室温下放置3-6h,40-60℃下干燥3-6h,100-120℃下干燥6-12h。
[0024]
s2:用含氮气体处理样品一,得到样品二。
[0025]
可选地,所述含氮气体为nh3、no、no2和n2o的一种。
[0026]
进一步地,所述处理温度为600-800℃,升温速率为0.5-2℃/mi2,处理空速为3000-6000h-1
处理时间为4-8h;满足所述含氮气氛下升温处理条件是在载体上引入碱性位,超出以上条件会造成处理不充分引起碱性位浓度不足或碱性位过量而占据了加氢活性位。
[0027]
s3:将样品二和碳酸钠溶液同时加入到过渡金属化合物水溶液中,待过渡金属化合物沉积于样品二上,将沉淀物过滤,干燥,得到样品三。
[0028]
进一步地,所述碳酸钠溶液的质量浓度为10-20%;满足所述碳酸钠的浓度范围才能使过渡金属充分沉淀,偏离此比例会造成过渡金属沉淀不足,不能有效形成金属碳化物前躯体。
[0029]
可选地,所述过渡金属化合物为硝酸镍、硝酸钴或钼酸铵中的一种。
[0030]
进一步地,所述过渡金属化合物与载体的质量比为 (1.91-10.14):(10-14);满足所述过渡金属化合物与载体的质量比可使金属化合物高度分散于载体表面,低于此比例形成的碳化物有效组分不足,而高于此比例则碳化物的分散性降低。
[0031]
进一步地,所述干燥为多次干燥;优选地,所述多次干燥为室温下放置3-6h,40-60℃下干燥3-6h,100-120℃下干燥6-12h。
[0032]
s4:将样品三在含碳气氛下升温处理,然后降至室温,用贫氧空气钝化,即得。
[0033]
进一步地,所述在含碳气氛下升温处理是以0.5-2.0℃/mi2的速率升温至400-700℃,处理时间为4-6h;气体空速为3000-6000h-1
;满足所述含碳气氛下升温处理条件是将碳化物前躯体还原为金属碳化物的最佳条件,超出以上条件会造成还原不足引起碳化物生成量不足或过度碳化而引起活性组分团聚。
[0034]
进一步地,所述用贫氧空气钝化是用o2含量为0.5-2.0%的贫氧空气钝化3-5h;满足所述钝化条件是将碳化物钝化的最佳条件,超出以上条件会造成催化剂无效钝化,接触空气后氧化以及造成催化剂钝化过度而生成大量氧化物。
[0035]
优选地,所述含碳气体为碳四以下烷烃或co中的一种。
[0036]
本发明的催化剂制备方法采用碳化的方法将金属ni、mo、co活化为金属碳化物,将助剂ceo2、la2o3等固载于载体上,并在载体上引入碱性位。金属碳化物、碱性位以及助剂三者的协同作用使得催化剂性能得以充分发挥,高选择性合成丁二酸酐。
[0037]
本发明还提供一种所述合成丁二酸酐的金属碳化物催化剂的应用:将所述催化剂置于固定床反应器内,在氢气空速400-600h-1
、压力3.0-6.0mpa 下,以0.5-2.0℃/mi2的速率升温至反应温度80-200℃,将顺酐溶解于溶剂中形成质量浓度为10-50%的溶液,以顺酐溶液体积空速1.0-10h-1
进料进行反应。
[0038]
可选地,所述溶剂为邻苯二甲酸二丁酯、1,4-二氧六环、四氢呋喃、γ
‑ꢀ
丁内酯或甲苯中的任意一种或多种的混合物。
[0039]
在固定床反应器中,采用该反应条件可有效发挥催化剂性能,可高选择性生成丁二酸酐。
[0040]
与现有技术相比,本发明将金属碳化物用于顺酐加氢合成丁二酸酐,催化剂具有耐酸腐蚀性,通过载体引入碱性位调节催化剂表面酸碱性性抑制富马酸的生成,通过引入助剂ceo2、la2o3促进碳化物和载体的协同作用。本发明催化剂中金属碳化物、碱性位以及助剂三者的协同作用使得催化剂具有高效顺酐加氢合成丁二酸酐性能。
具体实施方式
[0041]
下面将结合具体实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0042]
本发明如对各物质的添加量没有特殊的限定,则采用任意配比均可。
[0043]
实施例1
[0044]
将0.49g硝酸铈加入到10ml去离子水中,浸渍于13g活性炭上,室温下放置4h,40℃干燥3h,110℃干燥12h,得到样品一。
[0045]
将样品一在nh3气氛下以0.5/mi2的速率升温至600℃,在此温度下处理5h,nh3空速为6000h-1
,降温后取出,得到样品二。
[0046]
称取硝酸镍5.22g加入到100ml去离子水中形成溶液一,将样品二和 10ml 12%的碳酸钠溶液同时加入到溶液一中。将得到的沉淀过滤洗涤,在室温下放置5h,30℃干燥6h,110℃干燥6h,得到样品三。
[0047]
将样品三在ch4气氛下以0.5/mi2的速率升温至500℃,在此温度下处理5h,ch4空速为6000h-1
,降温后用o2含量为1%的贫氧空气钝化5h。
[0048]
所得催化剂碳化镍百分含量7.9wt%、氧化铈百分含量1.1wt%、载体活性炭百分含量91.0wt%,催化剂碱性位浓度为0.36mmol/g。
[0049]
将上述催化剂2ml装填于固定床管式反应器内。将催化剂在氢气空速 600h-1
、压力5.0mpa下,以1.0℃/mi2升高到200℃,以40%的顺酐/1,4 二氧六环溶液为反应原料,液体空速5.0h-1
进行加氢反应。反应结果列于表1中。
[0050]
实施例2
[0051]
将0.50g硝酸镧加入到10ml去离子水中,浸渍于12g氧化钛上,室温下放置5h,40℃干燥4h,120℃干燥8h,得到样品一。
[0052]
将样品一在no气氛下以1/mi2的速率升温至800℃,在此温度下处理 6h,气体空速为4000h-1
,降温后取出,得到样品二。
[0053]
称取硝酸钴5.52g加入到100ml去离子水中形成溶液一,将样品二和 10ml 10%的碳酸钠溶液同时加入到溶液一中。将得到的沉淀过滤洗涤,在室温下放置5h,50℃干燥3h,120℃干燥9h,得到样品三。
[0054]
将样品三在co气氛下以0.5/mi2的速率升温至400℃,在此温度下处理6h,气体空速为4000h-1
,降温后用o2含量为0.5%的贫氧空气钝化5h。
[0055]
所得催化剂碳化钴百分含量9.2wt%、氧化镧百分含量1.2wt%、载体氧化钛含量89.6wt%,催化剂碱性位浓度为0.33mmol/g。
[0056]
将上述催化剂2ml装填于固定床管式反应器内。将催化剂在氢气空速500h-1
、压力3.0mpa,以2.0℃/mi2升高到170℃,以30%的顺酐/四氢呋喃溶液为反应原料,液体空速10.0h-1
进行加氢反应。反应结果列于表 1中。
[0057]
实施例3
[0058]
将0.16g硝酸铈加入到10ml去离子水中,浸渍于10g二氧化硅上,室温下放置3h,60℃干燥3h,100℃干燥6h,得到样品一。
[0059]
将样品一在no2气氛下以0.5/mi2的速率升温至750℃,在此温度下处理4h,气体空速为5000h-1
,降温后取出,得到样品二。
[0060]
称取钼酸铵1.91g加入到100ml去离子水中形成溶液一,将样品二和10ml 14%的碳酸钠溶液同时加入到溶液一中。将得到的沉淀过滤洗涤,在室温下放置3h,60℃干燥3h,100℃干燥6h,得到样品三。
[0061]
将样品三在乙烷气氛下以1/mi2的速率升温至600℃,在此温度下处理 4h,气体空速为5000h-1
,降温后用o2含量为1.5%的贫氧空气钝化3h。
[0062]
所得催化剂碳化钼百分含量10.4wt%、氧化铈百分含量0.5wt%、载体二氧化硅含量89.1wt%,催化剂碱性位浓度为0.25mmol/g。
[0063]
将上述催化剂2ml装填于固定床管式反应器内。将催化剂在氢气空速 400h-1
、压力4.0mpa,以1.5℃/mi2升高到140℃,以20%的顺酐/γ-丁内酯溶液为反应原料,液体空速1.0h-1
进行加氢反应。反应结果列于表1中。
[0064]
实施例4
[0065]
将0.15g硝酸镧加入到10ml去离子水中,浸渍于14g氧化锆上,室温下放置6h,50℃干燥6h,120℃干燥10h,得到样品一。
[0066]
将样品一在n2o气氛下以2/mi2的速率升温至700℃,在此温度下处理 8h,气体空速为3000h-1
,降温后取出,得到样品二。
[0067]
称取硝酸镍10.14g加入到100ml去离子水中形成溶液一,将样品二和 10ml 16%的碳酸钠溶液同时加入到溶液一中。将得到的沉淀过滤洗涤,在室温下放置6h,50℃干燥6h,120℃干燥10h,得到样品三。
[0068]
将样品三在丙烷气氛下以2/mi2的速率升温至700℃,在此温度下处理 5h,气体空速为3000h-1
,降温后用o2含量为2%的贫氧空气钝化3h。
[0069]
所得催化剂碳化镍百分含量13.5wt%、氧化镧百分含量0.3wt%、载体氧化锆含量86.2wt%,催化剂碱性位浓度为0.45mmol/g。
[0070]
将上述催化剂2ml装填于固定床管式反应器内。将催化剂在氢气空速 500h-1
、压力5.0mpa,以2℃/mi2升高到120℃,以10%的顺酐/甲苯溶液为反应原料,液体空速7.0h-1
进行加氢反应。反应结果列于表1中。
[0071]
实施例5
[0072]
将0.29g硝酸铈加入到10ml去离子水中,浸渍于12g活性炭上,室温下放置3h,40℃干燥5h,100℃干燥9h,得到样品一。
[0073]
将样品一在氨气气氛下以1.5/mi2的速率升温至650℃,在此温度下处理7h,气体空速为4000h-1
,降温后取出,得到样品二。
[0074]
称取钼酸铵7.64g加入到100ml去离子水中形成溶液一,将样品二和 10ml 20%的碳酸钠溶液同时加入到溶液一中。将得到的沉淀过滤洗涤,在室温下放置3h,40℃干燥5h,100℃干燥9h,得到样品三。
[0075]
将样品三在丁烷气氛下以1.5/mi2的速率升温至500℃,在此温度下处理6h,气体空速为4000h-1
,降温后用o2含量为1%的贫氧空气钝化4h。
[0076]
所得催化剂碳化钼百分含量12.0wt%、氧化铈百分含量0.8wt%、载体活性炭含量87.2wt%,催化剂碱性位浓度为0.53mmol/g。
[0077]
将上述催化剂2ml装填于固定床管式反应器内。将催化剂在氢气空速 600h-1
、压力6.0mpa,以0.5℃/mi2升高到80℃,以50%的顺酐/邻苯二甲酸二丁酯溶液为反应原料,液体空速3.0h-1
进行加氢反应。反应结果列于表1中。
[0078]
对比例1
[0079]
将0.49g硝酸铈加入到10ml去离子水中,浸渍于13g活性炭上,室温下放置4h,40℃
干燥3h,110℃干燥12h,得到样品一。
[0080]
称取硝酸镍5.22g加入到100ml去离子水中形成溶液一,将样品一和 10ml 12%的碳酸钠溶液同时加入到溶液一中。将得到的沉淀过滤洗涤,在室温下放置5h,30℃干燥6h,110℃干燥6h,得到样品二。
[0081]
将样品二在ch4气氛下以0.5/mi2的速率升温至500℃,在此温度下处理5h,ch4空速为6000h-1
,降温后用o2含量为1%的贫氧空气钝化5h。
[0082]
所得催化剂碳化镍百分含量7.6wt%、氧化铈百分含量1.0wt%、载体活性炭百分含量91.4wt%,催化剂碱性位浓度为0.05mmol/g。
[0083]
将上述催化剂2ml装填于固定床管式反应器内。将催化剂在氢气空速 600h-1
、压力5.0mpa下,以1.0℃/mi2升高到200℃,以40%的顺酐/1,4 二氧六环溶液为反应原料,液体空速5.0h-1
进行加氢反应。反应结果列于表1中。
[0084]
对比例2
[0085]
将0.15g硝酸镧加入到10ml去离子水中,浸渍于14g氧化锆上,室温下放置6h,50℃干燥6h,120℃干燥10h,得到样品一。
[0086]
称取硝酸镍10.14g加入到100ml去离子水中形成溶液一,将样品一和 10ml 16%的碳酸钠溶液同时加入到溶液一中。将得到的沉淀过滤洗涤,在室温下放置6h,50℃干燥6h,120℃干燥10h,得到样品二。
[0087]
将样品二在丙烷气氛下以2/mi2的速率升温至700℃,在此温度下处理 5h,气体空速为3000h-1
,降温后用o2含量为2%的贫氧空气钝化3h。
[0088]
所得催化剂碳化镍百分含量13.7wt%、氧化镧百分含量0.3wt%、载体氧化锆含量86.0wt%,催化剂碱性位浓度为0.03mmol/g。
[0089]
将上述催化剂2ml装填于固定床管式反应器内。将催化剂在氢气空速 500h-1
、压力5.0mpa,以2℃/mi2升高到120℃,以10%的顺酐/甲苯溶液为反应原料,液体空速7.0h-1
进行加氢反应。反应结果列于表1中。
[0090]
对比例3
[0091]
将10g二氧化硅在no2气氛下以0.5/mi2的速率升温至750℃,在此温度下处理4h,气体空速为5000h-1
,降温后取出,得到样品一。
[0092]
称取钼酸铵1.91g加入到100ml去离子水中形成溶液一,将样品一和 10ml 14%的碳酸钠溶液同时加入到溶液一中。将得到的沉淀过滤洗涤,在室温下放置3h,60℃干燥3h,100℃干燥6h,得到样品二。
[0093]
将样品二在乙烷气氛下以1/mi2的速率升温至600℃,在此温度下处理 4h,气体空速为5000h-1
,降温后用o2含量为1.5%的贫氧空气钝化3h。
[0094]
所得催化剂碳化钼百分含量10.5wt%、载体二氧化硅含量89.5wt%,催化剂碱性位浓度为0.29mmol/g。
[0095]
将上述催化剂2ml装填于固定床管式反应器内。将催化剂在氢气空速 400h-1
、压力4.0mpa,以1.5℃/mi2升高到140℃,以20%的顺酐/γ-丁内酯溶液为反应原料,液体空速1.0h-1
进行加氢反应。反应结果列于表1中。
[0096]
表1催化剂顺酐加氢合成丁二酸酐性能
[0097][0098]
由表1可见,采用实施例1-5的催化剂,合成丁二酸酐性能优异,无富马酸的形成,且液相产物中未检测到过渡金属,说明催化剂具有良好的耐腐蚀性。而由对比例1-3的结果可见,加氢产品丁二酸酐选择性变差,且有富马酸形成;其液相产物中也未检测到过渡金属,说明由于金属碳化物的存在,催化剂仍具有耐酸腐蚀性。
[0099]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。此外,本领域的技术人员能够理解,尽管在此的一些实施例包括其它实施例中所包括的某些特征而不是其它特征,但是不同实施例的特征的组合意味着处于本发明的范围之内并且形成不同的实施例。例如,在上面的权利要求书中,所要求保护的实施例的任意之一都可以以任意的组合方式来使用。公开于该背景技术部分的信息仅仅旨在加深对本发明的总体背景技术的理解,而不应当被视为承认或以任何形式暗示该信息构成已为本领域技术人员所公知的现有技术。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1