一种提高Pd(OH)2/C催化剂氢解脱苄活性的方法
一种提高pd(oh)2/c催化剂氢解脱苄活性的方法
技术领域
1.本发明涉及一种提高pd(oh)2/c催化剂氢解脱苄活性的方法及其应用,属于催化剂制备技术领域。
技术背景
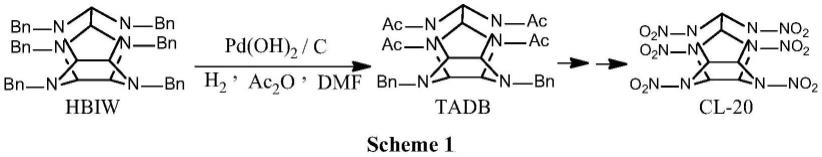
2.苄基是有机合成中重要的胺基、羟基保护基团。苄基的引入通常较为容易,但苄基的脱除有时却让人感到很棘手,尤其是n-苄基的脱除。六苄基六氮杂异伍兹烷(hbiw)是合成含能材料六硝基六氮杂异伍兹烷(cl-20)的主要前驱体,因此,hbiw的氢解脱苄是合成cl-20的关键步骤(scheme 1)。但由于hbiw笼形母体的稳定性较差,脱苄反应只能在较温和条件下进行,对催化剂性能提出更高要求。
[0003][0004]
钯炭催化剂(pd/c或pd(oh)2/c)几乎成为氢解脱苄反应的首选催化剂(j.mol.catal.a:chem.1996,112,437)。大量研究表明钯炭催化剂的性能与炭载体的本征性质,如孔结构、表面化学性质等密切相关(j.mol.catal.a:chem.2001,173,75;j.catal.2019,372,226)。研究发现以具有介孔结构的sibunit、cmk-3及“carbo medicinalis”活性炭为载体制备的pd/c催化剂具有较高的脱苄活性,而以微孔碳(norit-sx)为载体的催化剂则基本无活性(react.kinet.catal.lett.2007,92,293;j.energ.mater.2017,35,251)。我们前期研究也发现具有介孔结构、总孔体积较大且微孔体积所占比例较小的活性炭是hbiw脱苄催化剂的适宜载体(含能材料,2014,22(4),441)。还发现炭载体表面适当浓度含氧官能团的存在有助于提高炭载体的亲水性,增强pd物种与炭载体间的相互作用,进而提高pd分散度、脱苄活性及稳定性(catalysts 2021,11,441)。
[0005]
表面修饰是调节炭材料表面结构和化学性质的有效途径。常见的表面修饰方法有物理修饰法和化学修饰法。表面物理修饰法是通过吸附、涂敷、包覆等物理手段对炭材料表面进行改性(材料导报,2006,(s1),5);表面化学修饰法是通过修饰剂分子与炭材料表面原子发生化学反应,而改变其表面结构和状态的方法,如:酯化法、偶联法(物理化学学报,2003,19(7),621)、微乳液法(火炸药学报,2002,(1),33)、氧化还原法(carbon,2003,41,1979)等。专利cn108330312b公开了一种用金属包覆层改性石墨烯的方法,制得了一种金属包覆石墨烯的复合材料,改善了石墨烯与金属基体间的润湿性,有利于其在金属基体中的分散,从而提高增强效果;专利cn103723716b则公开了一种利用水热碳化制备氮掺杂碳包覆氧化石墨烯二维复合材料的方法,所得复合材料具有良好的导电性能。
[0006]
本发明提供了一种提高pd(oh)2/c催化剂氢解脱苄活性的方法,即采用氧化活化和氮掺杂碳包覆有机结合的方法对商业活性炭进行表面改性,以这种活性炭@水热碳复合材料为载体制备的pd(oh)2/c催化剂氢解脱苄活性得到显著提高。氧化活化和水热氮掺杂
碳包覆有机结合,既调节了活性炭的孔结构和表面化学性质,又改善了pd物种与炭载体间的相互作用,提高了pd分散度。按照本发明方法合成的pd(oh)2/c催化剂在hbiw氢解脱苄反应中催化活性得到显著提高,远优于以商业活性炭为载体的同类催化剂。
技术实现要素:
[0007]
根据现有技术存在的缺陷,本发明的目的之一是提供一种活性炭@水热碳复合材料的制备方法;本发明的目的之二是提供一种提高pd(oh)2/c催化剂在hbiw氢解脱苄反应中催化活性的方法。
[0008]
为实现上述发明目的,本发明采用的技术方案如下:
[0009]
一种提高pd(oh)2/c催化剂氢解脱苄活性的方法,其特征在于,包括以下步骤:
[0010]
(1)将商业活性炭加入到一定质量浓度的氧化剂溶液中进行氧化活化,然后用蒸馏水洗涤,烘干备用;
[0011]
(2)将步骤(1)中氧化活化后的活性炭与有机碳源、有机氮源及水加入到高压反应釜中,在一定搅拌转速、一定温度下进行水热处理;
[0012]
(3)将步骤(2)中得到的黑色固体在管式炉中焙烧,温度为500~1000℃,升温速率为1~20℃/min,保温时间1~8h,气氛为惰性气体;
[0013]
(4)将步骤(3)中得到的产物浸渍于质量浓度为5~37%的hcl溶液中,搅拌1~10h,用蒸馏水洗涤至中性,烘干备用,得到活性炭@水热碳复合材料载体;
[0014]
(5)称取一定量步骤(4)中得到的活性炭@水热碳复合材料载体分散于一定量蒸馏水/乙醇溶液中,根据负载量计算钯前驱体用量,将钯前驱体用盐酸溶解,倒入上述悬浮液中,搅拌一定时间,使钯离子在载体表面充分吸附,然后控制体系温度在15℃以下,用碱溶液调节体系ph至9~11,过滤,并充分洗涤至滤液检不出氯离子为止;产物在60℃下烘干备用。
[0015]
上述步骤(1)中,氧化剂为过氧化氢溶液或硝酸溶液中的至少一种。氧化剂溶液质量浓度为5%~40%,优选浓度为10%~30%;优选处理时间为2~4h;洗涤控制ph值为4-6。
[0016]
上述步骤(2)中,有机碳源为葡萄糖、蔗糖、果糖等中的至少一种;氮源为三聚氰胺、尿素、乌洛托品等中的至少一种。
[0017]
上述步骤(2)中,活性炭与有机碳源的质量比例为1:0.5~1:4,优选为1:1~1:2。活性炭与有机氮源的质量比例为1:0.5~1:4,优选为1:1~1:2;
[0018]
上述步骤(2)中水热反应条件为:温度150~240℃,优选为170~190℃;水热时间12~24h,优选为16-20h;搅拌速率50~400r/min,优选为100~200r/min;
[0019]
上述步骤(3)中,焙烧温度为500~1000℃,优选为700~900℃;升温速率为1~20℃/min,优选为5~10℃/min;惰性气体选自氮气、氦气或氩气中的至少一种,惰性气体流速为50~100ml/min;
[0020]
上述步骤(4)中,hcl的质量浓度为5~37%,优选为10~30%。搅拌时间为1~10h;
[0021]
上述步骤(5)中,炭载体与蒸馏水/乙醇溶液的质量比为1:5~1:50(每1g载体对应1-10ml蒸馏水/乙醇溶液),优选为1:10~1:20。蒸馏水/乙醇溶液的体积比为5:1~25:1。钯前驱体溶液与碳载体悬浮液的混合时间为0.5~2h。所用碱为naoh、na2co3及nahco3中的至少一种,其优选质量浓度为5-15%。
[0022]
钯的负载量为2-12wt%,优选8wt%。
[0023]
本发明与其他现有技术相比较具有以下特点
[0024]
(1)本发明采用氧化活化和氮掺杂碳包覆有机结合的方法对商业活性炭进行表面改性,提供了一种新的活性炭表面性质和孔结构调控方法。
[0025]
(2)以本发明制备的活性炭@水热碳复合材料为载体,采用优化的沉积沉淀法制备pd(oh)2/c催化剂,其在hbiw氢解脱苄反应中催化活性较未改性的商业活性炭有显著提高。
[0026]
实施例中选用两种商业活性炭(macklin公司的催化剂载体专用活性炭,8-16目;acros organics公司的颗粒状活性炭,20-40目)对本发明做具体说明。两种商业活性炭分别记作mc、ac;经氧化处理后,分别记作mco、aco;以mco、aco为基体,以葡萄糖为碳源,尿素为氮源,经水热处理并焙烧后,所得活性炭@水热碳复合材料分别记作mco-g-u、aco-g-u(其中g代表葡萄糖,u代表尿素);以未经氧化处理的ac为基体,相同水热条件下所得对比样品记作ac-g-u。以经氧化处理的aco为基体,仅以葡萄糖为碳源,经水热处理并焙烧后,所得的未掺氮活性炭@水热碳复合材料对比样品记作aco-g。
[0027]
mco、aco及mc
o-g-u和ac
o-g-u的扫描电镜照片如图1所示。可见氧化处理的活性炭样品与活性炭@水热碳复合材料样品的形貌很相似,而且水热包覆并焙烧后样品质量有明显增加,且增加量与有机碳源在相似条件下的碳化率相当,表明在活性炭表面形成了水热碳包覆层。mco、aco及mc
o-g-u和ac
o-g-u的氮气等温吸脱附曲线和织构数据分别如图2和表1所示。可以看出活性炭@水热碳复合材料样品的比表面积较基体活性炭有显著降低,表明氮掺杂碳包覆层堵塞了部分基体活性炭的孔道,尤其是微孔孔道。mco和aco的微孔占比分别为87.8%和54.2%,改性后微孔占比分别降为75.7%和45.2%。微孔占比的降低对提高相应催化剂hbiw氢解脱苄活性是有利的。透射电镜(tem)分析结果表明(图3),pd(oh)2/mc
o-g-u和pd(oh)2/ac
o-g-u催化剂上pd的分散度明显高于pd(oh)2/mco和pd(oh)2/aco。
[0028]
实施例中制备的pd(oh)2/c催化剂在hbiw氢解脱苄反应中的活性数据如表3所示。对于pd(oh)2/mco催化剂,在钯用量为底物质量0.5%时,基本无活性,得不到氢解产物。而对于pd(oh)2/mc
o-g-u催化剂,在钯用量为底物质量0.2%时,氢解反应可以顺利完成,产物收率高达89.4%。同样,对于pd(oh)2/aco催化剂,在钯用量为底物质量0.2%时,基本无活性。但对于pd(oh)2/ac
o-g-u催化剂,即使钯用量降为底物质量0.15%时,氢解反应仍可以顺利完成,产物收率89.3%。表明以活性炭@水热碳复合材料为载体制备的pd(oh)2/c催化剂在hbiw氢解脱苄中活性得到了大幅度提高。对比催化剂pd(oh)2/ac-g-u和pd(oh)2/ac
o-g在不同钯用量下的活性结果表明(表2),在未经氧化处理的基体活性炭表面直接引入氮掺杂包覆层或在氧化处理的基体活性炭表面引入未氮掺杂的水热碳包覆层,对提高相应催化剂的氢解脱苄活性都有一定贡献。但pd(oh)2/ac-g-u和pd(oh)2/ac
o-g催化剂的活性均低于pd(oh)2/ac
o-g-u,表明基体活性炭的氧化活化和水热碳包覆层的掺氮杂化对提高催化剂氢解脱苄活性都是有益的。但只有当将基体活性炭的氧化活化和氮掺杂碳包覆有机结合时,所得催化剂在hbiw氢解脱苄反应中的活性最佳。
附图说明
[0029]
图1为实施例1中经氧化处理的活性炭样品mco(a)及氮掺杂碳包覆后的载体mc
o-g-u(b)和实施例2中经氧化处理的活性炭样品aco(c)及氮掺杂碳包覆后的载体ac
o-g-u(d)的
扫描电镜(sem)照片。
[0030]
图2为催化剂的tem照片及其粒径分布图:pd(oh)2/mco(a,b)、pd(oh)2/mco-g-u(c,d)、pd(oh)2/aco(e,f)及pd(oh)2/aco-g-u(g,f)。
[0031]
图3为经氧化处理的活性炭样品mco和aco,氮掺杂碳包覆后载体mc
o-g-u、和ac
o-g-u及实施例5和6中制备的对比样品ac-g-u和ac
o-g的氮气等温吸脱附曲线。
具体实施方式
[0032]
下面结合实施例对本发明内容作进一步说明,但本发明不限于以下实施例。
[0033]
实施例1:
[0034]
称取mc活性炭20g于三颈烧瓶中,加入200ml的水和400ml质量分数为30%的过氧化氢溶液,在60℃水浴下氧化处理4h,洗涤至ph=4-6,烘干得到mco。称取葡萄糖10g和尿素10g溶于250ml水中,得到葡萄糖、尿素混合溶液。称取5g mco倒入上述混合溶液中,室温下搅拌30min。然后将混合溶液倒入高压反应釜中进行水热反应。水热温度180℃,水热时间12h,搅拌速率为200r/min。将所得的黑色粉末用蒸馏水和酒精洗涤,烘干备用。将上述得到的黑色粉末研磨后转入瓷舟,置于管式炉中800℃焙烧2h。用10wt%hcl浸泡2h,过滤、洗涤、干燥,得到mc
o-g-u样品。
[0035]
称取mc
o-g-u样品4.0g,加入到40ml纯水中,并加入2ml乙醇。称量0.538g氯化钯,用1.2ml浓盐酸将其溶解,并用适量蒸馏水稀释。将氯化钯溶液倒入上述碳载体悬浮液中,室温搅拌1h。然后控制体系温度在15℃以下,用naoh溶液调节体系ph至9~11。过滤,并充分洗涤至滤液检不出氯离子为止。产物在60℃下烘干,得到催化剂pd(oh)2/mc
o-g-u。
[0036]
实施例2:
[0037]
按照实例1中相同的方法处理商业活性炭ac,分别得到aco和ac
o-g-u载体样品。
[0038]
以ac
o-g-u为载体,按照实例1中相似的方法制备催化剂,得到催化剂pd(oh)2/ac
o-g-u。
[0039]
实施例3:
[0040]
以mco为载体,按照实例1中相似的方法制备催化剂,得到催化剂pd(oh)2/mco。
[0041]
实施例4
[0042]
以aco为载体,按照实例1中相似的方法制备催化剂,得到催化剂pd(oh)2/aco。
[0043]
实施例5
[0044]
称取葡萄糖10g和尿素10g溶于250ml水中,得到葡萄糖、尿素混合溶液。称取5g商业活性炭ac倒入上述混合溶液中,室温下搅拌30min。然后将混合溶液倒入高压反应釜中进行水热反应。水热温度180℃,水热时间12h,搅拌速率为200r/min。将所得的黑色粉末用蒸馏水和酒精洗涤,烘干备用。将上述得到的黑色粉末研磨后转入瓷舟,置于管式炉中800℃焙烧2h。用10wt%hcl浸泡2h,过滤、洗涤、干燥,得到对比载体ac-g-u。
[0045]
以ac-g-u为载体,按照实例1中相似的方法制备催化剂,得到催化剂pd(oh)2/ac-g-u。
[0046]
实施例6
[0047]
称取葡萄糖10g溶于250ml水中,得到葡萄糖溶液。称取5g经氧化活化处理的基体活性炭商业活性炭aco倒入上述混合溶液中,室温下搅拌30min。然后将混合溶液倒入高压
反应釜中进行水热反应。水热温度180℃,水热时间12h,搅拌速率为200r/min。将所得的黑色粉末用蒸馏水和酒精洗涤,烘干备用。将上述得到的黑色粉末研磨后转入瓷舟,置于管式炉中800℃焙烧2h。用10wt%hcl浸泡2h,过滤、洗涤、干燥,得到对比载体ac
o-g。
[0048]
以aco-g为载体,按照实例1中相似的方法制备催化剂,得到催化剂pd(oh)2/aco-g。
[0049]
催化剂活性评价条件
[0050]
以hbiw的氢解脱苄乙酰化为探针反应,评价所得催化剂的活性。具体测试条件如下:称取hbiw 50g,按照pd的设计用量计算催化剂的加入量,反应介质为dmf(100ml)和ac2o(50ml)组成的混合体系,同时,加入助剂溴苯0.9ml。加完料后,抽真空,用氢气置换3次,然后通入氢气,开动搅拌,常温常压下反应一定时间,待体系析出固体氢解产物后,升温至40℃,继续反应。总反应时间约20h。过滤洗涤干燥,得氢解脱苄粗产物。
[0051]
表1为经氧化处理的活性炭样品mco和aco,氮掺杂碳包覆后载体mc
o-g-u、和ac
o-g-u及实施例5和6中制备的对比样品ac-g-u和ac
o-g的比表面积、平均孔径及孔体积。
[0052]
表1
[0053][0054]
表2为实施例1,2,3,4,5,6中所制备的催化剂pd(oh)2/mco-g-u、pd(oh)2/aco-g-u、pd(oh)2/mco、pd(oh)2/aco、pd(oh)2/ac-g-u、pd(oh)2/aco-g在hbiw氢解脱苄反应中的活性数据。
[0055]
表2
[0056][0057]
*nr(no reaction):基本无活性,即吸氢量较少,hbiw氢解脱苄反应难完成,得不到固体产物。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1