一种耐溶剂复合纳滤膜及其制备方法和应用
1.本发明涉及纳滤膜技术领域,具体涉及一种耐溶剂复合纳滤膜及其制备方法和应用。
背景技术:
2.纳滤膜是介于反渗透膜和超滤膜之间的一种压力驱动的新型分离膜,相较于反渗透膜具有操作压力低、渗透通量高的优点;与超滤膜相比,可截留分子量在200~1000道尔顿的物质,分离性能高,在较低的操作压力下能够具有较高的渗透通量。纳滤技术已广泛的应用于水分离纯化、纺织印染、医药和食品化工等领域。但是随着工业中有机溶剂的使用越来越多,对环境和人类的日常生活也产生了一定的威胁,因此对有机溶剂的分离和纯化引起了许多人的关注。开发一种具有优良分离性能和耐溶剂性的纳滤膜,替代传统的、耗能高的、具有污染性的分离方式具有重要的意义。
3.目前,耐溶剂纳滤膜主要分为两种:(1)整体皮层不对称膜:主要由相转化法一步制的,由上表面致密的皮层和底部多孔的支撑层组成,整体皮层不对称膜的皮层的厚度和孔径对分离效果起到了关键性的作用,但是此类膜较厚,溶剂的渗透通量较低。(2)薄膜复合膜:主要是由界面聚合法制的,由支撑层超滤基膜和较薄的分离层组成,例如,karan等(karan s,jiang z,livingston a g.membrane filtration.sub-10nm polyamide nanofilms with ultrafast solvent transport for molecular separation[j].science(new york,n.y.),2015,348(6241):1347-1351.)采用在交联p84超滤膜上引入氢氧化镉纳米线,纳米线溶解形成粗糙的表面和空腔结构;gohil等(gohil j m,ray p.polyvinyl alcohol as the barrier layer in thin film composite nanofiltration membranes:preparation,characterization,and performance evaluation[j].journal of colloid&interface science,2009,338(1):121-127.)通过在聚砜超滤膜表面涂敷聚乙烯醇水溶液,以马来酸作为交联剂,制备出了交联聚乙烯醇薄膜复合纳滤膜;rajaeian等(rajaeian b,rahimpour a,tade m o,et al.fabrication and characterization of polyamide thin film nanocomposite(tfn)nanofiltration membrane impregnated with tio2nanoparticles[j].desalination,2013,313(complete):176-188.)采用二氧化钛做水相溶液的添加剂,制备了聚酰胺薄膜纳米复合膜。
[0004]
但是上述薄膜复合膜具有典型的trade-off效应,即渗透通量和截留率呈此消彼长的状态。
技术实现要素:
[0005]
有鉴于此,本发明的目的在于提供一种耐溶剂复合纳滤膜及其制备方法和应用,本发明提供的耐溶剂复合纳滤膜的截留率高且渗透通量高。
[0006]
为了实现上述发明目的,本发明提供以下技术方案:
[0007]
本发明提供了一种耐溶剂复合纳滤膜,包括聚酰亚胺超滤支撑膜和位于所述聚酰亚胺超滤支撑膜表面的聚酰胺分离层;
[0008]
所述聚酰胺分离层包括聚酰胺基层和掺杂在所述聚酰胺基层中的纳米粒子接枝环糊精,所述纳米粒子接枝环糊精中的纳米粒子包括纳米凹凸棒土和纳米二氧化钛中的至少一种。
[0009]
优选地,所述聚酰胺分离层的厚度为50~300nm;
[0010]
所述聚酰胺分离层中纳米粒子接枝环糊精的掺杂量为0.05~0.25wt%;
[0011]
所述纳米粒子接枝环糊精中环糊精的接枝率为3~7%。
[0012]
优选地,所述聚酰亚胺超滤支撑膜厚度为50~300μm。
[0013]
本发明提供了上述技术方案所述耐溶剂复合纳滤膜的制备方法,包括以下步骤:
[0014]
(1)将聚酰亚胺、致孔剂和极性溶剂混合,将得到的聚酰亚胺铸膜液成膜后在水中相转化,得到聚酰亚胺超滤支撑膜;
[0015]
(2)将所述聚酰亚胺超滤支撑膜与交联剂溶液混合,进行交联反应,得到交联支撑膜;
[0016]
(3)将所述交联支撑膜与纳米粒子接枝环糊精与有机胺类水相单体和水混合,进行浸渍,得到含饱和水相单体支撑膜;
[0017]
(4)将所述含饱和水相单体支撑膜置于酰氯类有机相单体溶液中,进行界面聚合反应形成聚酰胺分离层,得到纳滤膜前驱体;
[0018]
(5)将所述纳滤膜前驱体进行溶剂活化,得到所述耐溶剂复合纳滤膜。
[0019]
优选地,步骤(1)中,所述聚酰亚胺和致孔剂的质量比为18~22:1~5;
[0020]
所述相转化的时间为1~5天。
[0021]
优选地,步骤(2)中,所述交联剂溶液的浓度为110~130g/l;
[0022]
所述交联剂溶液中的交联剂为有机胺类化合物;
[0023]
所述交联反应的温度为20~30℃,时间为12~18h。
[0024]
优选地,步骤(3)中,所述纳米粒子接枝环糊精和有机胺类水相单体的质量比为0.05~0.25:2~3;
[0025]
所述有机胺类水相单体包括间苯二胺、邻苯二胺、对苯二胺、三聚氰胺、硫脲、聚乙烯亚胺和二乙基三胺和n-氨基乙基哌嗪中的至少一种。
[0026]
优选地,步骤(4)中,所述酰氯类有机相单体溶液中酰氯类有机相单体包括邻苯二甲酰氯、对苯二甲酰氯、间苯二甲酰氯、均苯三甲酰氯,5-异氰酸酯-异酞酰氯和5-氧甲酰氯-异酞酰氯中的至少一种;
[0027]
所述界面聚合反应的温度为20~30℃,时间为30~60s。
[0028]
优选地,步骤(5)中,所述溶剂活化用溶剂包括酰胺类溶剂和吡咯烷酮类溶剂中的至少一种;
[0029]
所述溶剂活化的温度为80~90℃,时间为10~60min。
[0030]
本发明提供了上述技术方案所述的耐溶剂复合纳滤膜或上述技术方案所述制备方法制备得到的耐溶剂复合纳滤膜在染料分离、有机溶剂分离或药物分离中的应用。
[0031]
本发明提供了一种耐溶剂复合纳滤膜,包括聚酰亚胺超滤支撑膜和位于所述聚酰亚胺超滤支撑膜表面的聚酰胺分离层;所述聚酰胺分离层包括聚酰胺基层和掺杂在所述聚
酰胺基层中的纳米粒子接枝环糊精,所述纳米粒子接枝环糊精中的纳米粒子包括纳米凹凸棒土和纳米二氧化钛中的至少一种。环糊精分子略呈锥形空心环状立体结构,外腔大量的亲水羟基基团,内部含有疏水基团,亲水结构可为极性溶剂提供通道,疏水结构可为非极性溶剂提供传输通道,并且,凹凸棒土具有管状结构,为溶剂的通过提供额外的传输通道,使得耐溶剂复合纳滤膜在保持高截留率的状态下提高膜的通量;纳米二氧化钛表面含有羟基基团,能够提高膜的亲水性,进而提高耐溶剂复合纳滤膜的极性溶剂的通量,通过利用环糊精对纳米粒子进行改性得到的纳米粒子接枝环糊精表面含有大量的羟基,使其能够很好的分散在聚酰胺分离层中,不易团聚,大大提高了聚酰胺分离层的亲水性,增加聚酰胺分离层的自由体积,突破trade-off效应,进而在保证截留率的情况下提高了渗透通量,使得耐溶剂复合纳滤膜同时具有高通量和高截留率,使得耐溶剂复合纳滤膜在分离染料方面具有很好的应用前景。
[0032]
进一步地,本发明提供的耐溶剂复合纳滤膜的聚酰胺分离层较薄,大大减少溶剂的传输阻力,提高耐溶剂复合纳滤膜的溶剂的渗透通量,使得耐溶剂纳滤膜同时具有高通量和高截留率。
[0033]
本发明提供了上述技术方案所述耐溶剂复合纳滤膜的制备方法。本发明通过聚酰亚胺超滤支撑膜与交联剂进行交联反应,能够提高耐溶剂性复合纳滤膜的耐溶剂性能;在制备聚酰胺分离层过程中加入纳米粒子接枝环糊精,可以在保持截留率相对稳定的状态下,有效提高耐溶剂复合纳滤膜的通量。本发明采用界面聚合方式制备聚酰胺分离层,纳米粒子接枝环糊精表面含有大量羟基,将其溶于有机胺类水相单体中,能够与有机胺类水相单体中的氨基(-nh2)之间形成氢键,减缓水相单体向酰氯类有机相扩散的速率,进而有助于形成较薄的聚酰胺分离层,减少溶剂的传输阻力,提高耐溶剂复合纳滤膜的溶剂的渗透通量,使耐溶剂纳滤膜具有高通量和高截留率。而且,本发明提供的制备方法操作简单,生产成本低,耗能低,绿色环保,适宜工业化生产。
附图说明
[0034]
图1为atp和实施例1制备的atp@β-cd的红外光谱图;
[0035]
图2为二氧化钛接枝β-环糊精的红外谱图;
[0036]
图3为添加不同浓度atp@β-cd纳米粒子后,耐溶剂复合纳滤膜的甲醇通量和伊文思蓝染料截留率测试结果图;
[0037]
图4为实施例6和对比例1~2制备的耐溶剂复合纳滤膜的的甲醇通量和伊文思蓝染料截留率测试结果图。
具体实施方式
[0038]
本发明提供了一种耐溶剂复合纳滤膜,包括聚酰亚胺超滤支撑膜和位于所述聚酰亚胺超滤支撑膜表面的聚酰胺分离层。在本发明中,所述聚酰胺分离层包括聚酰胺基层和掺杂在所述聚酰胺基层中的纳米粒子接枝环糊精,所述纳米粒子接枝环糊精中的纳米粒子包括纳米凹凸棒土和纳米二氧化钛中的至少一种;所述纳米粒子接枝环糊精中环糊精的接枝率优选为3~7%,更优选为4~6%,进一步优选为5%;所述环糊精优选包括α-环糊精、β-环糊精和γ-环糊精中的至少一种。在本发明中,所述聚酰胺分离层中纳米粒子接枝环糊精
的掺杂量优选为0.05~0.25wt%,更优选为0.05~0.2wt%。在本发明中,所述聚酰胺分离层的厚度优选为50~300nm,更优选为100~250nm。在本发明中,所述聚酰亚胺超滤支撑膜厚度为优选50~300μm,更优选100~250μm。
[0039]
本发明提供了上述技术方案所述耐溶剂复合纳滤膜的制备方法,包括以下步骤:
[0040]
(1)将聚酰亚胺、致孔剂和极性溶剂混合,将得到的聚酰亚胺铸膜液成膜后在水中相转化,得到聚酰亚胺超滤支撑膜;
[0041]
(2)将所述聚酰亚胺超滤支撑膜与交联剂溶液混合,进行交联反应,得到交联支撑膜;
[0042]
(3)将所述交联支撑膜与纳米粒子接枝环糊精与有机胺类水相单体和水混合,进行浸渍,得到含饱和水相单体支撑膜;
[0043]
(4)将所述含饱和水相单体支撑膜置于酰氯类有机相单体溶液中,进行界面聚合反应形成聚酰胺分离层,得到纳滤膜前驱体;
[0044]
(5)将所述纳滤膜前驱体进行溶剂活化,得到所述耐溶剂复合纳滤膜。
[0045]
在本发明中,若无特殊说明,所有的原料组分均为本领域技术人员熟知的市售商品。
[0046]
本发明将聚酰亚胺、致孔剂和极性溶剂混合,将得到的聚酰亚胺铸膜液成膜后在水中相转化,得到聚酰亚胺超滤支撑膜。
[0047]
在本发明中,所述致孔剂优选包括聚乙二醇、聚乙烯吡咯烷酮和羟丙基纤维素中的至少一种。在本发明中,所述聚酰亚胺和致孔剂的质量比优选为18~22:1~5,更优选为18.5~21.5:1~4,进一步优选为19~21:1~3,最优选为20:1。在本发明中,所述极性溶剂优选包括n,n-二甲基甲酰胺、n,n-二甲基乙酰胺和n-甲基吡咯烷酮中的至少一种。在本发明中,所述聚酰亚胺铸膜液中聚酰亚胺的浓度优选为18~22%,更优选为19~21%,进一步优选为20%;所述聚酰亚胺铸膜液中致孔剂的浓度优选为1~5%,更优选为2~4%,进一步优选为3%。
[0048]
在本发明中,所述混合的方式优选为搅拌混合,所述混合的温度优选为室温,所述混合的时间优选为24~48h,更优选为24~30h。完成所述混合后,本发明优选还包括静置,所述静置的温度优选为室温,所述静置的时间优选为1~7天,更优选为2~3天,所述静置的目的是除去混合所得体系中的气泡,提高聚酰亚胺超滤支撑膜的均匀性。
[0049]
在本发明中,所述成膜优选为:将聚酰亚胺铸膜液涂覆在载体表面进行成膜。在本发明中,所述载体优选为无纺布,所述无纺布的材质优选包括聚对苯二甲酸乙二醇酯、丙纶、锦纶、粘胶纤维、腈纶、乙纶或氯纶;所述载体的孔径优选为10~150μm,更优选为50~100μm。在本发明中,所述涂覆优选为浇注,所述浇注优选采用浇注刀进行,所述浇注刀的尺寸优选为100~350μm,更优选为150~300μm,进一步优选为200~250μm;所述浇注速度优选为0.025~0.05m/s,更优选为0.025~0.045m/s,进一步优选为0.025~0.04m/s;本发明对于所述浇注的时间没有特殊限定,以得到厚度为50~300μm的聚酰亚胺超滤支撑膜为准。
[0050]
在本发明中,所述相转化的温度优选为室温,所述相转化优选包括依次进行第一相转化和第二相转化,具体的,将聚酰亚胺铸膜液成膜后置于水中进行第一相转化,然后置于新的水中进行第二相转化;所述水优选为去离子水;所述第一相转化的温度优选为室温;所述第一相转化的时间优选为10~60min,更优选为10~20min,所述第一相转化过程中析
出聚酰亚胺超滤支撑膜以及大量的dmf;所述第二相转化的时间优选为1~5天,更优选为2~3天;所述第二相转化过程中继续析出聚酰亚胺超滤支撑膜。
[0051]
完成所述相转化后,本发明优选还包括将所得相转化膜进行有机溶剂洗涤,得到聚酰亚胺超滤支撑膜。在本发明中,所述有机溶剂洗涤用有机溶剂优选为醇类溶剂,更优选包括异丙醇、乙醇和丙醇中的至少一种;所述有机溶剂洗涤的次数优选为3~4次,单次有机溶剂洗涤的时间优选为40~60min,更优选为60min;所述有机溶剂洗涤的目的是清除聚酰亚胺超滤支撑膜中残余的水和溶剂。
[0052]
得到聚酰亚胺超滤支撑膜后,本发明将所述聚酰亚胺超滤支撑膜与交联剂溶液混合,进行交联反应,得到交联支撑膜。
[0053]
在本发明中,所述交联剂溶液的浓度优选为110~130g/l,更优选为115~125g/l,进一步优选为120g/l;所述交联剂溶液中的交联剂优选为有机胺类化合物,更优选包括乙二胺、丁二胺、己二胺、4,4'-二氨基二苯醚、间苯二胺、二乙基三胺和超支化聚乙烯亚胺中的至少一种;所述交联剂溶液中的溶剂优选为醇类溶剂,更优选包括异丙醇、乙醇和丙醇中的至少一种。
[0054]
在本发明中,所述交联反应的温度优选为20~30℃,更优选为20~25℃;所述交联反应的时间优选为12~18h,更优选为14~16h;本发明通过交联能够提高耐溶剂性复合纳滤膜的耐溶剂性能。
[0055]
完成所述交联反应后,本发明优选还包括将所得交联反应膜进行溶剂置换,得到交联支撑膜。在本发明中,所述溶剂置换用溶剂优选为醇类溶剂,更优选包括异丙醇、乙醇和丙醇中的至少一种;所述溶剂置换的次数优选为3~4次,单次溶剂置换的时间优选为40~60min,更优选为60min;所述溶剂置换的目的是清除交联支撑膜中残余的交联剂。
[0056]
得到交联支撑膜后,本发明将所述交联支撑膜与纳米粒子接枝环糊精与有机胺类水相单体和水混合,进行浸渍,得到含饱和水相单体支撑膜。
[0057]
在本发明中,所述纳米粒子接枝环糊精的制备方法优选包括以下步骤:
[0058]
将纳米粒子进行酸化处理,得到酸化纳米粒子;所述纳米粒子包括凹凸棒土(atp)和二氧化钛中的至少一种;
[0059]
将所述酸化纳米粒子与硅烷偶联剂、催化剂和有机溶剂混合,进行脱醇缩聚反应,得到硅烷化纳米粒子;
[0060]
将所述硅烷化纳米粒子与环糊精、活化剂和无水极性非质子溶剂混合,进行接枝反应,得到纳米粒子接枝环糊精。
[0061]
本发明纳米粒子进行酸化处理,得到酸化纳米粒子;所述纳米粒子包括凹凸棒土(atp)和二氧化钛中的至少一种。
[0062]
在本发明中,所述纳米粒子的粒径优选为500~2000nm,更优选为500~1500nm。在本发明中,所述酸化处理用酸优选包括盐酸、硝酸和硫酸中的至少一种,所述酸的浓度优选为1~5mol/l,更优选为2~4;酸化时间为4~6h,酸化方式为混合搅拌。在本发明中,所述纳米粒子的质量和酸物质的量之比优选为1g:4~30mmol,更优选为1g:5~25mmol,进一步优选为1g:10~20mmol。
[0063]
在本发明中,所述酸化处理的温度优选为室温,所述酸化处理的时间优选为4~6h,更优选为5h;所述酸化处理优选在搅拌条件下进行。在本发明中,所述酸化处理的目的
是去除纳米粒子中的杂质,使其孔道疏松;而且,由于凹凸棒土的等电点为3,二氧化钛的等电点为3.6,通常情况下羟基脱氢带负电荷,对纳米粒子进行酸处理能够使纳米粒子中更多的羟基结构显现出来,便于后续的接枝改性。
[0064]
完成所述酸化处理后,本发明优选还包括将所得酸化处理液依次进行固液分离,将所得固体组分进行干燥,得到酸化纳米粒子。本发明对于所述固液分离方式没有特殊限定,采用本领域技术人员熟知的固液分离方式即可,具体如过滤。在本发明中,所述干燥的温度优选为80~120℃,更优选为100℃,所述酸化处理的时间优选为4~6h,更优选为5h。
[0065]
得到酸化纳米粒子后,本发明将所述酸化纳米粒子与硅烷偶联剂、催化剂和有机溶剂混合,进行脱醇缩聚反应,得到硅烷化纳米粒子。
[0066]
在本发明中,所述混合优选为搅拌混合,在本发明的具体实施例中,所述混合优选为将所述酸化纳米粒子分散于有机溶剂中,然后加入硅烷偶联剂和催化剂混合。在本发明中,所述硅烷偶联剂优选包括乙烯基三乙氧基硅烷(a151)、乙烯基三甲氧基硅烷(a171)、γ-氨丙基三乙氧基硅烷(kh540)、γ-氨丙基三乙氧基硅烷(kh550)、γ-缩水甘油醚氧丙基三甲氧基硅烷(kh560)和γ-甲基丙烯酰氧基丙基三甲氧基硅烷(kh570)中的至少一种。在本发明中,所述纳米粒子和硅烷偶联剂的质量比优选为4~5:1,更优选为4.5:1。在本发明中,所述催化剂优选包括吡啶和三乙胺中的至少一种。在本发明中,所述纳米粒子的质量和催化剂的体积之比优选为6g:0.5~1ml,更优选为6g:0.6~0.8ml。在本发明中,所述有机溶剂优选包括甲苯和二甲苯中的至少一种,所述有机溶剂优选为无水有机溶剂。在本发明中,所述纳米粒子的质量和有机溶剂的体积之比优选为6g:100~300ml,更优选为6g:100~200ml。
[0067]
在本发明中,所述脱醇缩聚反应的温度优选为80~100℃,更优选为90℃;所述脱醇缩聚反应的时间优选为24~36h,更优选为25~30h;所述脱醇缩聚反应优选在保护气氛下进行,所述保护气氛优选为惰性气体,更优选为氩气或氩气;以凹凸棒土和kh560为例,所述脱醇缩聚反应过程中发生的反应如式(1)所示:
[0068][0069]
完成所述脱醇缩聚反应后,本发明优选还包括将所得脱醇缩聚反应液冷却至室温后固液分离,将所得固体产物进行洗涤后干燥,得到硅烷化纳米粒子。本发明对于所述冷却没有特殊限定,采用本领域技术人员熟知的冷却方式即可,具体如自然冷却。本发明对于所述固液分离没有特殊限定,采用本领域技术人员熟知的固液分离方式即可,具体如过滤。在本发明中,所述洗涤优选包括依次进行苯类溶剂洗涤、醇类溶剂洗涤、水洗和酮类溶剂洗涤;所述苯类溶剂洗涤优选包括甲苯洗涤或二甲苯洗涤;所述醇类溶剂洗涤优选为乙醇洗涤;所述酮类溶剂洗涤优选为丙酮洗涤;所述苯类溶剂洗涤、醇类溶剂洗涤、水洗和丙酮洗涤的次数独立地优选为3~4次。在本发明中,所述干燥的温度优选为60~80℃,本发明对于所述干燥的时间没有特殊限定,干燥至恒重即可,具体如8~10h。
[0070]
得到硅烷化纳米粒子后,本发明将所述硅烷化纳米粒子与环糊精与活化剂和无水极性非质子溶剂混合,进行接枝反应,得到纳米粒子接枝环糊精。
[0071]
在本发明中,所述环糊精在使用前优选先进行重结晶。在本发明中,所述重结晶用溶剂优选为水。在本发明中,所述环糊精与水的质量比优选为1:5~7.5,更优选为1:5.5~
7。在本发明中,所述重结晶优选为:将环糊精溶解于水中,在室温条件下进行析晶,固液分离,将所得晶体进行干燥,得到重结晶环糊精。在本发明中,所述溶解的温度优选为90~100℃,更优选为95℃。本发明对于所述固液分离没有特殊限定,采用本领域技术人员熟知的固液分离方式即可,具体如过滤。在本发明中,所述干燥的温度优选为90~100℃,本发明对于所述干燥的时间没有特殊限定,干燥至恒重即可,具体如8~10h。
[0072]
在本发明中,所述硅烷化纳米粒子与环糊精的质量比优选为1:0.5~1.0,更优选为1:0.6~0.8。
[0073]
在本发明中,所述活化剂优选包括氢化钠和氢化钾中的至少一种。在本发明中,所述环糊精与活化剂的质量比优选为10~15:1,更优选为10~13:1。在本发明中,所述极性非质子溶剂优选包括n,n-二甲基甲酰胺、n,n-二甲基乙酰胺和n-甲基吡咯烷酮中的至少一种。在本发明中,所述纳米粒子的质量与极性非质子溶剂的体积之比优选为6g:100~150ml,更优选为6g:120~150ml。
[0074]
在本发明中,所述混合优选为搅拌混合;所述混合的温度优选为室温;在本发明的具体实施例中,所述混合优选为:将环糊精溶解于无水极性非质子溶剂中,加入活化剂混合至无气体排出,固液分离,将所得液体组分与硅烷化纳米粒子混合。本发明对于所述固液分离没有特殊限定,采用本领域技术人员熟知的固液分离方式即可,具体如过滤。
[0075]
在本发明中,所述接枝反应的温度优选为110~120℃,更优选为115℃;所述接枝反应的时间优选为12~18h,更优选为12~15h;所述接枝反应优选在搅拌和保护气氛下进行,所述保护气氛优选为惰性气体,更优选为氩气或氩气。在本发明中,以式(1)所示反应的产物和氢化钠为例,所述活化和接枝反应过程中发生的反应如式(2)所示:
[0076][0077]
完成所述述接枝反应后,本发明优选还包括将所得接枝产物进行洗涤后干燥,得到纳米粒子接枝环糊精。在本发明中,所述洗涤优选包括依次进行极性非质子溶剂洗涤、醇洗和水洗;所述极性非质子溶剂洗涤用极性非质子溶剂优包括n,n-二甲基甲酰胺、n,n-二甲基乙酰胺和n-甲基吡咯烷酮中的至少一种;所述醇洗优选为甲醇洗;所述极性非质子溶剂洗涤、醇洗和水洗的次数独立地优选为3~4次。在本发明中,所述干燥的温度优选为优选为60~80℃,本发明对于所述干燥的时间没有特殊限定,干燥至恒重即可,具体如8~10h。
[0078]
在本发明中,所述有机胺类水相单体优选包括间苯二胺、邻苯二胺、对苯二胺、三聚氰胺、硫脲、聚乙烯亚胺和二乙基三胺和n-氨基乙基哌嗪中的至少一种。在本发明中,所述纳米粒子接枝环糊精和有机胺类水相单体的质量比优选为0.05~0.25:2~3,更优选为0.05~0.2:2~2.8,进一步优选为0.1~0.15:2~2.5。
[0079]
在本发明中,所述交联支撑膜与纳米粒子接枝环糊精、有机胺类水相单体和水混合的混合温度优选为室温;在本发明的具体实施例中,所述混合优选为将纳米粒子接枝环糊精、有机胺类水相单体和水超声混合,得到纳米粒子接枝环糊精-水相单体混合液;将所述交联支撑膜置于所述纳米粒子接枝环糊精-水相单体溶液中。在本发明中,所述纳米粒子接枝环糊精-水相单体混合液中纳米粒子接枝环糊精的质量浓度优选为0.05~0.25%,更
优选为0.05~0.2%;所述纳米粒子接枝环糊精-水相单体混合液中有机胺类水相单体的浓度优选为2~3%,更优选为2~2.8%,进一步优选为2~2.5%。在本发明中,所述超声混合的温度优选为室温,所述超声混合的时间优选为20~40min,更优选为20~30min。本发明对于所述纳米粒子接枝环糊精-水相单体混合液的用量没有特殊限定,能够将交联支撑膜浸没即可。
[0080]
在本发明中,所述浸渍的温度优选为20~25℃,所述浸渍的时间优选为2~10min,更优选为2~5min。
[0081]
完成所述浸渍后,本发明优选还包括将所得浸渍膜中残留的纳米粒子接枝环糊精-水相单体混合液去除,得到含饱和水相单体支撑膜;所述去除优选利用橡胶辊进行。
[0082]
得到含饱和水相单体支撑膜后,本发明将所述含饱和水相单体支撑膜置于酰氯类有机相单体溶液中,进行界面聚合反应形成聚酰胺分离层,得到所述纳滤膜前驱体。
[0083]
在本发明中,所述酰氯类有机相单体溶液中酰氯类有机相单体优选包括邻苯二甲酰氯、对苯二甲酰氯、间苯二甲酰氯、均苯三甲酰氯,5-异氰酸酯-异酞酰氯和5-氧甲酰氯-异酞酰氯中的至少一种;所述酰氯类有机相单体溶液中酰氯类有机相单体的质量浓度优选为0.01~0.02%,更优选为0.01~0.015%;所述酰氯类有机相单体溶液中的溶剂优选包括正己烷和正戊烷中的至少一种。
[0084]
在本发明中,所述界面聚合反应的温度优选为20~30℃,更优选为25℃,所述界面聚合反应的时间优选为30~60s,更优选为40~60s。在本发明中,所述界面聚合反应过程中,含饱和水相单体支撑膜中的有机胺类水相单体与酰氯类有机相单体发生界面聚合反应形成聚酰胺分离层。
[0085]
完成所述界面聚合反应后,本发明优选还包括将所得复合膜进行洗涤后静置,得到纳滤膜前驱体。在本发明中,所述洗涤用溶剂优选包括正己烷和正戊烷中的至少一种;所述洗涤的次数优选为2~3次;所述洗涤的目的是除去未反应的酰氯类有机相单体。在本发明中,所述静置优选为置于空气中静置,所述静置的时间优选为2~5min,更优选为3~4min。
[0086]
得到纳滤膜前驱体后,本发明将所述纳滤膜前驱体进行溶剂活化,得到所述耐溶剂复合纳滤膜。
[0087]
在本发明中,所述溶剂活化用溶剂优选包括酰胺类溶剂和吡咯烷酮类溶剂中的至少一种,更优选包括n,n-二甲基甲酰胺、n,n-二甲基乙酰胺和n-甲基吡咯烷酮中的至少一种;本发明对于所述溶剂的用量没有特殊限定,能够将所述纳滤膜前驱体浸没即可。在本发明中,所述溶剂活化的温度优选为80~90℃,更优选为80~85℃;所述溶剂活化的时间优选为10~60min,更优选为10~20min;所述溶剂活化的目的是除去分子量较小的聚酰胺,为溶剂提供更多的通路,进而提高耐溶剂复合纳滤膜的渗透通量。在本发明中,所述耐溶剂复合纳滤膜优选储存于溶剂中,所述溶剂优选包括醇类溶剂,更优选包括甲醇、乙醇和丙醇中的至少一种。
[0088]
所述溶剂活化后,本发明优选还包括:将所述溶剂活化得到的溶剂活化复合膜进行溶质交换,得到耐溶剂复合纳滤膜。在本发明中,所述溶质交换优选为将溶剂活化复合膜浸泡于甲醇中进行溶质交换,所述溶质交换的温度优选为室温,所述溶质交换的时间优选为15~20min;所述溶质交换的目的是除去酰胺类溶剂。
[0089]
在本发明中,所述耐溶剂复合纳滤膜优选保存于甲醇中。
[0090]
本发明提供了上述技术方案所述的耐溶剂复合纳滤膜或上述技术方案所述制备方法制备得到的耐溶剂复合纳滤膜在染料分离、有机溶剂分离或药物分离中的应用。
[0091]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0092]
实施例1
[0093]
(1)凹凸棒土接枝β-环糊精的制备
[0094]
将10g凹凸棒土(atp,粒径为500~1500nm)置于浸于40ml浓度为3mol/l的盐酸中,在室温条件下酸化处理5h,过滤,将所得固体组分在100℃条件下真空干燥12h,得到酸化凹凸棒土。
[0095]
将6g酸化凹凸棒土悬浮于150ml干燥的甲苯中,加入1.5gγ-缩水甘油醚氧丙基三甲氧基硅烷(kh560)和1ml三乙胺混合均匀,在氩气气氛、90℃条件下脱醇缩聚反应24h,降至室温下,过滤,将所得固体组分甲苯洗涤3次,无水乙醇洗涤3次,蒸馏水洗涤3次,丙酮洗涤3次,在60℃条件下真空干燥8h,得到硅烷化凹凸棒土(命名为si-atp)。
[0096]
将8gβ-环糊精(β-cd)置于50ml的蒸馏水中,加热至95℃,待β-环糊精完全溶解后,转移到室温下进行析晶,将析出的晶体过滤后置于烘箱中,在100℃条件下干燥8h,得到重结晶β-环糊精。
[0097]
将3g重结晶β-环糊精溶于无水dmf中,加入0.3g氢化钠混合均匀,在室温条件下搅拌至不再排放气体,搅拌时间为30min,过滤,在所得液体组分中加入5g硅烷化凹凸棒土,在110℃、氮气气氛下搅拌12h,dmf洗涤3次,甲醇洗涤3次,蒸馏水洗涤3次,在60℃条件下真空干燥8h,得到凹凸棒土接枝β-环糊精(命名为atp@β-cd)。
[0098]
(2)耐溶剂复合纳滤膜的制备
[0099]
将聚酰亚胺p84溶于dmf中,加入聚乙烯吡咯烷酮(pvp),室温下搅拌24h,静置2天,采用250μm浇铸刀,将所得聚酰亚胺铸膜液(p84浓度为20wt%,pvp的浓度为1wt%)以0.025m/s的速度浇注在孔径为23.31μm的聚对苯二甲酸乙二醇酯(pet)无纺布上,然后浸入到去离子水中,在室温条件下相转化10min,然后转移到新的去离子水中继续相转化2天,然后浸泡于异丙醇中溶剂置换4次(单次溶剂置换时间为60min),得到聚酰亚胺超滤支撑膜(厚度约为200μm)。
[0100]
将所述聚酰亚胺超滤支撑膜浸泡在1,6-己二胺的异丙醇溶液(1,6-己二胺浓度为120g/l)中,在室温条件下交联16h,浸泡于异丙醇中洗涤4次(单次洗涤时间为60min),得到交联支撑膜,贮存在水浴中备用。
[0101]
将所述凹凸棒土接枝β-环糊精、间苯二胺和水在室温条件下超声20min,得到25ml凹凸棒土接枝β-环糊精-水相单体混合液(间苯二胺的质量浓度为2.0%,凹凸棒土接枝β-环糊精的质量浓度为0.05%)。将所述交联支撑膜浸入到所述凹凸棒土接枝β-环糊精-水相单体混合水液中,在室温条件下浸渍2min,然后用橡胶辊去除膜表面残留的溶液,在空气中自然晾干,得到含饱和水相单体支撑膜。
[0102]
将所述含饱和水相单体支撑膜浸入到均苯三甲酰氯的正己烷溶液(均苯三甲酰氯
浓度为0.01wt%)中,在室温条件下界面聚合60s,用正己烷冲洗3次,在含饱和水相单体支撑膜表面形成聚酰胺分离层(厚度为200~300nm),将得到的纳滤膜前驱体浸泡于80℃的dmf中活化30min,然后浸于甲醇中,在室温下条件下溶质交换15min,得到耐溶剂复合纳滤膜(命名为tfn-0.05膜),置于甲醇中保存备用。
[0103]
图1为atp和atp@β-cd的红外光谱图,由图1可知,在3620cm-1
,3554cm-1
和1034cm-1
处出现atp的特性峰,其中3620cm-1
和3554cm-1
处的特征峰为al-oh和fe-oh的伸缩振动峰,1034cm-1
处是atp中si-o-si振动吸收峰;atp@β-cd红外谱图在2935cm-1
、2874cm-1
处出现两个新的特征峰,这是由于β-cd上的c-h吸收振动峰的影响,此外在1652cm-1
和1471cm-1
处的吸收峰有所增强,这是由于β-cd中的-oh与β-cd中的-ch2伸缩振动导致峰变强。表明,β-cd成功负载在atp上。
[0104]
实施例2
[0105]
按照实施例1的方法制备耐溶剂复合纳滤膜,与实施例1的区别仅在于:凹凸棒土接枝β-环糊精-水相单体混合水液中凹凸棒土接枝β-环糊精的质量浓度为0.10%,得到耐溶剂复合纳滤膜(命名为tfn-0.1膜)
[0106]
实施例3
[0107]
按照实施例1的方法制备耐溶剂复合纳滤膜,与实施例1的区别仅在于:凹凸棒土接枝β-环糊精-水相单体混合水液中凹凸棒土接枝β-环糊精的质量浓度为0.15%,得到耐溶剂复合纳滤膜(命名为tfn-0.15膜)。
[0108]
实施例4
[0109]
按照实施例1的方法制备耐溶剂复合纳滤膜,与实施例1的区别仅在于:凹凸棒土接枝β-环糊精-水相单体混合水液中凹凸棒土接枝β-环糊精的质量浓度为0.20%,得到耐溶剂复合纳滤膜(命名为tfn-0.20膜)。
[0110]
实施例5
[0111]
按照实施例1的方法制备耐溶剂复合纳滤膜,与实施例1的区别仅在于:凹凸棒土接枝β-环糊精-水相单体混合水液中凹凸棒土接枝β-环糊精的质量浓度为0.25%,得到耐溶剂复合纳滤膜(命名为tfn-0.25膜)。
[0112]
实施例6
[0113]
按照实施例3的方法制备耐溶剂复合纳滤膜,与实施例3的区别仅在于:将凹凸棒土替换为粒径为100nm的二氧化钛,二氧化钛接枝β-环糊精的水相单体溶液中二氧化钛接枝β-环糊精(命名为tio2@β-cd)的质量浓度为0.15%,得到耐溶剂复合纳滤膜(命名为tfn-0.15tio2膜)。
[0114]
图2为二氧化钛接枝β-环糊精的红外谱图,由图2可知,在2923cm-1
处归属于β-环糊精中亚甲基伸缩振动峰,在1158cm-1
和1031cm-1
处的峰是因为二氧化钛纳米颗粒表面上物理吸附的水而导致二氧化钛八面体纳米晶中ti-o键的变形振动产生的,在940cm-1
处的峰与β-环糊精的结构单体吡喃糖中的c-o键伸缩振动有关,1647cm-1
处是吸附水的伸缩振动峰。
[0115]
对比例1
[0116]
按照实施例1步骤(2)的方法制备耐溶剂复合纳滤膜,与实施例1的区别仅在于:将步骤(2)中的凹凸棒土接枝β-环糊精替换为实施例1步骤(1)中的凹凸棒土,得到耐溶剂复合纳滤膜(命名为tfn-0.10t膜)。
[0117]
对比例2
[0118]
按照实施例1步骤(2)的方法制备耐溶剂复合纳滤膜,与实施例1的区别仅在于:步骤(2)中不添加凹凸棒土接枝β-环糊精,得到耐溶剂复合纳滤膜(命名为tfc膜)。
[0119]
测试例1
[0120]
采用20mg/l的伊文思蓝的甲醇溶液测试实施例1~6和对比例1~2制备的耐溶剂复合纳滤膜的纳滤性能,测试的操作压力为10bar,按式(1)计算渗透通量,按式(2)计算截留率。公式(一)和公式(二)如下:
[0121][0122]
式(1)中,a为有效膜面积(m2),δt为操作时间(h),δv为操作时间下渗透液的体积(l),δp为操作压力(bar);
[0123]
式(2)中,cf为原料液染料的浓度,c
p
为渗透液染料的浓度,截留率r的数值在0~100%之间,r的数值越大,说明透过的染料越少,分离性能越好。
[0124]
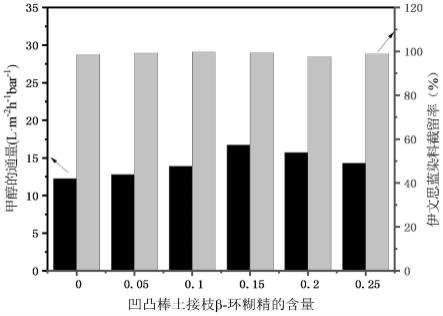
测试结果如表1和图3~4所示,其中,图3为添加不同浓度atp@β-cd纳米粒子后,耐溶剂复合纳滤膜的甲醇通量和伊文思蓝染料截留率测试结果图,图4为实施例6和对比例1~2制备的耐溶剂复合纳滤膜的的甲醇通量和伊文思蓝染料截留率测试结果图。
[0125]
表1耐溶剂复合纳滤膜的甲醇通量和伊文思蓝染料的截留率
[0126][0127]
由表1和图3~4可知,随着纳米粒子atp@β-cd浓度的增加,耐溶剂复合纳滤膜的甲醇通量和伊文思蓝染料截留也逐步提高,在纳米粒子atp@β-cd浓度为0.15w/v%时,溶剂甲醇的通量为16.71l/(m2·h·
bar),伊文思蓝染料(960.81da)的截留率为99.32%,而不加入任何纳米粒子的耐溶剂复合纳滤膜,溶剂甲醇的通量仅为12.25l/(m2·h·
bar),溶剂的通量增加了36.41%,同时伊文思蓝染料的截留也略有增加。这是因为纳米粒子atp@β-cd的加入为溶剂的渗透提供了额外的传输通道,且纳米粒子的加入阻碍了界面聚合的快速反应,有利于形成较致密较薄的聚酰胺分离层,进而减小溶剂的传输阻力,从而增大了耐溶剂复合纳滤膜的溶剂的渗透通量。
[0128]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1