2‑取代的苯并噻唑的电化学合成方法与流程
本发明涉及2-取代的苯并噻唑的合成领域,具体涉及2-取代的苯并噻唑的电化学合成。
背景技术:
取代的苯并噻唑是一类重要的药用杂环化合物;它们存在于许多生物活性分子如临床使用的药物唑泊(用于糖尿病的治疗),选择性脂肪酸酰胺水解酶抑制剂、醛糖还原酶抑制剂、抗肿瘤药物以及脂肪酸氧化抑制剂。此外,苯并噻唑骨架也经常存在于其它分子,如工业染料,天然产品,功能材料,农药,荧光指示剂以及反应的配体中。因此探索出一种快速高效的方法来构建苯并噻唑引起了科学家的广泛兴趣。
2-取代的苯并噻唑一般是在金属催化下,通过邻卤n-苯基硫代酰胺类化合物的环化反应或者苯并噻唑的2号位直接的c-h活化反应来构建。另一类方法是通过2-氨基苯硫酚与醇、醛(酮)与羧酸通过缩合反应来合成。这种方法通常需要处于强酸或者强氧化剂的条件下。近年来,huang小组成功的在电化学条件下,应用二价钴作为催化剂,2-氨基苯硫酚以及醇为原料,在恒流的单池中成功的构建了苯并噻唑类化合物,水对该反应有重要的影响(lai.yin.-long.;ye.jian.-shan.;huang.jing.-mei.chem.eur.j,2016,22,5425-5429)。同年,该课题组又成功的通过2-氨基苯硫酚与酮酸的缩合反应构建了2-取代苯并噻唑类化合物,该反应条件较为温和,但是需要同时添加酸与碱[wang.h.-b.;huang.j.-m.advancedsynthesis&catalysis,2016,358,1975-1981]。据我们所知,直接的通过2-氨基苯硫酚(或者2,2'-二氨基二苯二硫醚)与环醚反应构建2-取代的苯并噻唑还没有见报道过。
近年来,随着人们环保观念的普及,电化学作为一种绿色的化学合成方法越来越受到科学家们的关注。电化学作为一种绿色的能源,具有以下优势:(1)可持续能源的生产,例如太阳能电池,氢燃料电池以及存储系统。(2)化学产品的电化学合成,电子本质上是一种清洁的反应物,可以应用于实现氧化还原反应。(3)电化学反应过程的实时监测,相较于传统的化学分析,电化学能够为正在进行的化学反应提供快速,实时的反馈。(4)污染物的处理,通过电化学技术,可以处理一些传统化学以及生物技术难以处理污染物。并且电化学技术是廉价与安全的。
技术实现要素:
本发明是在传统有机化学方法合成2-取代的苯并噻唑的基础上利用2-氨基苯硫酚(或者2,2'-二氨基二苯二硫醚)与四氢呋喃在铁作为催化剂,电作为氧化剂的条件下合成2-取代的苯并噻唑的方法。以2-氨基苯硫酚(或者2,2'-二氨基二苯二硫醚)与四氢呋喃作为原料,采用惰性电极,在无隔膜单室电解池中加入电解质和三氯化铁以及水,常温常压恒电流条件下获得2-取代的苯并噻唑,本发明反应中的环醚作为反应的溶剂又作为反应的底物,反应无需添加质子酸或者碱,环境友好,条件温和,选择性好,产率较高,整个反应过程简单易行。
为了达到上述目的,本发明采用以下技术方案。
2-取代的苯并噻唑的电化学合成方法,包括如下步骤:
1)将电解质、反应底物、铁盐和水加入到电解溶剂中,插入电极,常温下搅拌并恒流反应;所述电解质为一水高氯酸钠或高氯酸锂,所述反应底物为2-氨基苯硫酚或2,2'-二氨基二苯二硫醚,所述铁盐为三氯化铁或四水合氯化亚铁;
2)反应完毕后,萃取、分离纯化,得到2-取代的苯并噻唑。
优选的,步骤1)所述电解溶剂为四氢呋喃或者四氢呋喃与乙腈的混合溶液。
优选的,步骤1)所述水的用量为500ul或600ul。
优选的,步骤1)所述电解质在电解液中的浓度为0.2mol/l-0.3mol/l,所述电解液为电解质、电解溶剂以及水的混合溶液。
优选的,步骤1)所述的电极中阴极和阳极的距离为10mm,阳极为φ10mm×15mm的rvc、镍、铜和铂片中的一种;阴极为φ10mm×15mm的铂片。
优选的,步骤1)所述反应的电流为8-12ma。
优选的,步骤1)所述的反应的时间为3-4小时。
优选的,步骤1)所述2-氨基苯硫酚与三氯化铁的物质的量之比为4:1。
优选的,步骤1)所述2,2'-二氨基二苯二硫醚与四水合氯化亚铁的物质的量之比为2:1。
优选的,以2-氨基苯硫酚为原料,本发明的合成路线原理如下:
以2-氨基苯硫酚和四氢呋喃为原料电化学制备2-取代的苯并噻唑的方法,包括以下步骤:在无隔膜的电解池中加入电解质、2-氨基苯硫酚、三氯化铁、电解溶剂、水,插入电极,搅拌,通电,10ma恒流条件下反应3.5h,反应完成后,用乙酸乙酯对电解液进行有机溶剂萃取,再分离提纯得到相应产物。
所述的电极中阴极为rvc、镍、铜、铂片的一种,阳极为铂片。
所述的电解溶剂为四氢呋喃或者四氢呋喃与乙腈的混合溶液。
所述的电解质为一水合高氯酸钠或高氯酸锂,电解质的摩尔浓度为0.2~0.3mol/l(相对于溶剂)。
所述的2-氨基苯硫酚与三氯化铁的摩尔比为4:1。
所述的水的量为500ul或600ul
本发明与现有技术相比具有如下优点:
(1)本发明第一次使用了环醚作为原料与2-氨基苯硫酚或2,2'-二氨基二苯二硫醚反应构建2-取代的苯并噻唑。
(2)本发明不需要添加质子酸碱与强氧化剂,使用绿色的水作为添加剂。体系简单,绿色环保。
(3)本发明避免了高温的苛刻条件,反应在常温常压下操作,简单、安全。
附图说明
图1为本发明实施例1制备的产物的1hnmr图谱。
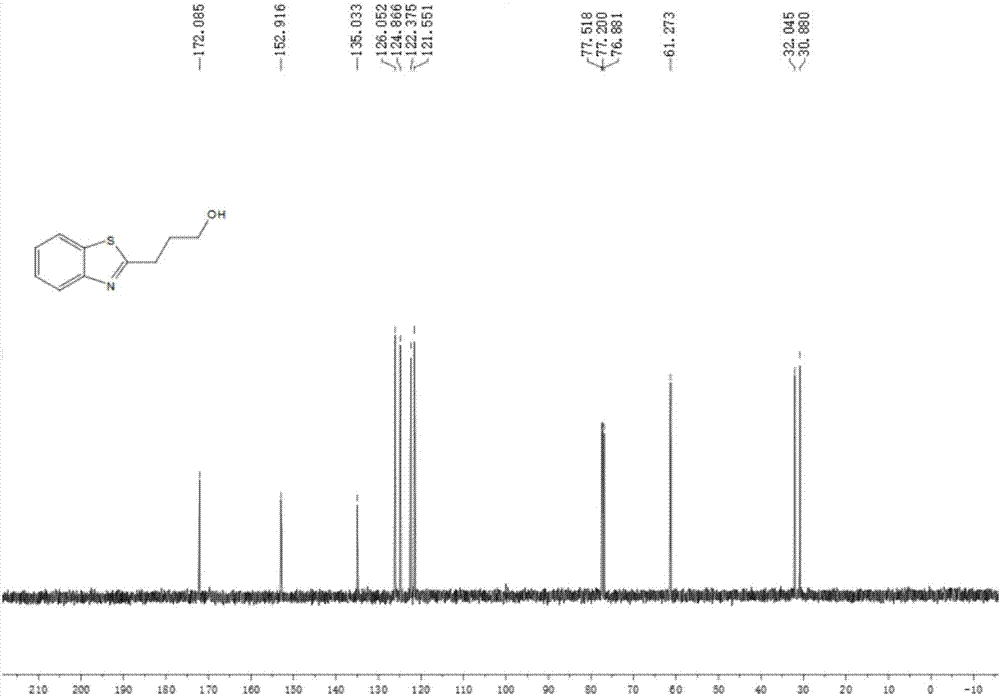
图2为本发明实施例1制备的产物的13cnmr图谱。
具体实施方式
下面结合实例对本发明作进一步详细的描述,但本发明要求保护的范围不限于此。
实施例1
以φ10mm×15mm的金属铂作为反应的阳极与阴极,在圆底烧瓶中依次加入1.6mmolnaclo4·h2o、0.0625mmolfecl3、0.25mmol2-氨基苯硫酚、5ml四氢呋喃、600ul水、磁力搅拌子,盖上盖子,接通电源,调节电流为10ma,常温下电解3.5h,反应结束后,用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和nacl水溶液(10ml×1)洗,无水na2so4干燥,减压蒸干,分离得产物3a,产率为68%。
所得产品的结果由1hnmr、13cnmr确定。
实施例2
以φ10mm×15mm的金属铂作为阳极,φ10mm×15mm的金属铜作为阴极,在圆底烧瓶中依次加入1.6mmolnaclo4·h2o、0.0625mmolfecl3、0.25mmol2-氨基苯硫酚、5ml四氢呋喃、600ul去离子水、磁力搅拌子,盖上盖子,接通电源,调节电流为10ma,常温下电解3.5h,反应结束后,用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和nacl水溶液(10ml×1)洗,无水na2so4干燥,减压蒸干,分离得产物3a,产率为53%。
所得产品的结果由1hnmr、13cnmr确定。
实施例3
以φ10mm×15mm的金属铂作为阳极,φ10mm×15mm的金属镍作为阴极,在圆底烧瓶中依次加入1.6mmolnaclo4·h2o、0.0625mmolfecl3、0.25mmol2-氨基苯硫酚、5ml四氢呋喃、600ul去离子水、磁力搅拌子,盖上盖子,接通电源,调节电流为10ma,常温下电解3.5h,反应结束后,用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和nacl水溶液(10ml×1)洗,无水na2so4干燥,减压蒸干,分离得产物3a,产率为50%。
所得产品的结果由1hnmr、13cnmr确定。
实施例4
以φ10mm×15mm的金属铂作为阳极,φ10mm×15mm的rvc作为阴极,在圆底烧瓶中依次加入1.6mmolnaclo4·h2o、0.0625mmolfecl3、0.25mmol2-氨基苯硫酚、5ml四氢呋喃、600ul去离子水、磁力搅拌子,盖上盖子,接通电源,调节电流为10ma,常温下电解3.5h,反应结束后,用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和nacl水溶液(10ml×1)洗,无水na2so4干燥,减压蒸干,分离得产物3a,产率为53%。
所得产品的结果由1hnmr、13cnmr确定。
实施例5
以φ10mm×15mm的金属铂同时作为反应的阳极与阴极,在圆底烧瓶中依次加入1.6mmolnaclo4·h2o、0.0625mmolfecl3、0.25mmol2-氨基苯硫酚、5ml四氢呋喃、500ul去离子水、磁力搅拌子,盖上盖子,接通电源,调节电流为10ma,常温下电解3.5h,反应结束后,用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和nacl水溶液(10ml×1)洗,无水na2so4干燥,减压蒸干,分离得产物3a,产率为60%。
所得产品的结果由1hnmr、13cnmr确定。
实施例6
以φ10mm×15mm的金属铂同时作为反应的阳极与阴极,在圆底烧瓶中依次加入1.6mmolnaclo4·h2o、0.0625mmolfecl3、0.25mmol2-氨基苯硫酚、3mlch3cn,2mlthf、600ul去离子水、磁力搅拌子,盖上盖子,接通电源,调节电流为10ma,常温下电解3.5h,反应结束后,用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和nacl水溶液(10ml×1)洗,无水na2so4干燥,减压蒸干,分离得产物3a,产率为62%。
所得产品的结果由1hnmr、13cnmr确定。
实施例7
以φ10mm×15mm的金属铂同时作为反应的阳极与阴极,在圆底烧瓶中依次加入1.6mmolnaclo4·h2o、0.0625mmolfecl3、0.25mmol2-氨基苯硫酚、2.5mlch3cn,2.5mlthf、600ul去离子水、磁力搅拌子,盖上盖子,接通电源,调节电流为10ma,常温下电解3.5h,反应结束后,用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和nacl水溶液(10ml×1)洗,无水na2so4干燥,减压蒸干,分离得产物3a,产率为64%。
所得产品的结果由1hnmr、13cnmr确定。
实施例8
以φ10mm×15mm的金属铂同时作为反应的阳极与阴极,在圆底烧瓶中依次加入1.6mmolnaclo4·h2o、0.0625mmolfecl3、0.25mmol2-氨基苯硫酚、2mlch3cn,3mlthf、600ul去离子水、磁力搅拌子,盖上盖子,接通电源,调节电流为10ma,常温下电解3.5h,反应结束后,用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和nacl水溶液(10ml×1)洗,无水na2so4干燥,减压蒸干,分离得产物3a,产率为62%。
所得产品的结果由1hnmr、13cnmr确定。
以2,2'-二氨基二苯二硫醚为原料,本发明的合成路线原理如下:
实施例9
以φ10mm×15mm的金属铂同时作为反应的阳极与阴极,在圆底烧瓶中依次加入1.6mmolnaclo4·h2o、0.125mmolfecl2·4h2o、0.25mmol2,2'-二氨基二苯二硫醚、5mlthf、600ul去离子水、磁力搅拌子,盖上盖子,接通电源,调节电流为10ma,常温下电解3.5h,反应结束后,用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和nacl水溶液(10ml×1)洗,无水na2so4干燥,减压蒸干,分离得产物3a,产率为83%。
所得产品的结果由1hnmr、13cnmr确定。
实施例10
以φ10mm×15mm的金属铂同时作为反应的阳极与阴极,在圆底烧瓶中依次加入1.3mmol、liclo4、0.125mmolfecl2·4h2o、0.25mmol2,2'-二氨基二苯二硫醚、5mlthf、600ul去离子水、磁力搅拌子,盖上盖子,接通电源,调节电流为10ma,常温下电解3.5h,反应结束后,用乙酸乙酯(15ml×3)对粗产物进行萃取,合并有机层,饱和nacl水溶液(10ml×1)洗,无水na2so4干燥,减压蒸干,分离得产物3a,产率为71%。
所得产品的结果由1hnmr、13cnmr确定。
分析实施例1得到的产物结构,并与masayasawamura[murakami.r.;iwai.t,.;sawamura.m.synlett,2016,27,1187-1192]报道的文献对照,结果证实该物质为具有1c结构式的产物。
产物1c的鉴定数据:
如图1所示,产物1c的1hnmr数据为:1hnmr(400mhz,cdcl3):δ(ppm)1hnmr(400mhz,cdcl3)δ:7.92(d,j=8.4hz,1h),7.78(d,j=8.0hz,1h),7.40(t,j=7.2hz,1h),7.30(t,j=7.6hz,1h),3.89(br,1h),3.75(t,j=6.0hz,2h),3.22(t,j=7.2hz,2h),2.11(m,2h)。
如图2所示,产物1c的13cnmr数据为:13cnmr(100mhz,cdcl3):δ(ppm)172.1,152.9,135.0,126.0,124.9,122.4,121.6,661.3,32.0,30.9。
- 还没有人留言评论。精彩留言会获得点赞!