达原饮组合物的质量控制方法与流程
本发明属于重要分析领域,具体涉及达原饮组合物的质量控制方法。
背景技术:
中药汤剂是临床用药的主流,但因起煎煮、携带不方便,煎煮制备的汤剂质量往往因人、因煎煮器具而存在较大差异,而经典名方临床疗效确切,因简、便、验、廉而深受广大消费者喜爱,运用现代科学技术将经典名方开发承携带方便、便于服用的中药制剂,不仅是对中医药的传承,也是更好的促进经典名方的临床应用。颗粒剂制备工艺相对简单、易于赋形,服用、携带方便,在服用形式上与传统汤剂最为一致,能较大限度的保持传统汤剂的特点,因此以汤剂形式服用的经典名方选择颗粒剂最为适宜。颗粒剂的开发应遵循“原汁原味”的原则,在现有技术条件下,首先通过研究制备明确煎煮参数的标准(基准)汤剂,对基准汤剂的关键质量属性进行研究,主要以单处方标准汤剂的固含量(干膏率)、指纹图谱、多指标成分作为质量参比,指导颗粒的研制。研究中为减少不同来源的饮片存在的质量差异对一致性研究带来影响,选择随行对照的方式,采用同一批次的饮片,煎煮至少10份标准汤剂,以其关键质量的均值作为颗粒剂制备工艺参数研制的“基准”。本发明采用指纹图谱技术,结合多波长切换技术测定多指标成分的方式对达原饮标准汤剂(以下称为达原饮组合物①)、达原饮标准颗粒(以下称为达原饮组合物②)的质量进行全面评价。
达原饮出自明代吴有性《瘟疫论》,由槟榔、厚朴、草果仁、知母、芍药、黄芩、甘草七味中药组成,主治瘟疫或疟疾,邪伏膜原证,现代临床常用疟疾、流行性感冒、病毒性脑炎,病毒性肝炎等属温热疫毒伏于膜原,湿热雍盛者等。该方为开达膜原、辟秽化浊之要方,方中厚朴散满、化痰下气、破戾气所结;草果辛烈辟秽,宣透伏邪;槟榔行气所结、使邪速渍。三药相伍,直达膜原—瘟疫盘结处,逐邪外出。瘟疫之邪内郁成熟,最易伤阴,故佐以黄芩清热,知母滋阴清热,芍药敛阴,用甘草可缓前三药之猛,又可制后三药之寒,诸药合用,开达膜原,辟秽化浊,热邪得清,邪去病解。达原饮现代临床应用广泛,在解热方面比抗菌,抗病毒药有更好的疗效。特别是对感染型发热和湿热型发热具有更重要的作用,且达原饮加减方在其它疾病治疗方面也运用广泛并有着确切的临床疗效,因此该方作为临床经典名方开发临床意义重大。
临床上应用“达原饮”治疗多种湿热病,均取得较好效果,该方是临床上的有效方剂,该方由7味药组成,其中:槟榔为棕榈科植物槟梆的干燥成熟种子,主要含有槟榔碱碱、槟榔次碱,及去甲基槟榔碱等生物碱,其水浸液对皮肤真菌,流感病毒有抑制作用。草果仁为姜科植物草果的干燥成熟果实,照清炒法炒至焦黄色并微鼓起,去壳,取仁所得,主要含有挥发油具有躁湿温中,除痰截疟的功效。厚朴为木兰科植物厚朴的干燥皮,根皮及枝皮,具有广谱的抗菌作用,对革兰氏阳性菌、耐酸性菌、肺炎球菌、白喉杆菌、溶血性链球菌、枯草杆菌、金黄色葡萄球菌、志贺氏及施氏痢疾杆菌及常见致病性皮肤真菌等均有抑制作用,厚朴酚、和厚朴酚、四氢厚朴酚及四氢和厚朴酚,均有极强的抑制细菌的作用。木兰醇、和厚朴酚等对TPA激活的Epstein-Barr病毒(Ebr)有明显的抑制作用。知母为百合科植物知母的干燥根茎,具有“滋阴”作用;对大肠杆菌所致家兔发热有解热作用,其解热有效成分为芒果苷;芒果苷还可镇咳、祛痰;知母对伤寒杆菌、痢疾杆菌、白喉杆菌、肺炎双球菌、葡萄球菌等均有一定抑制作用;异芒果苷还具有显著的抗单纯疱疹病毒作用,芒果苷和异芒果苷均可阻止HSV-I在细胞内的复制。黄芩为唇形科植物黄芩的干燥根,具有明显的抗炎作用,黄芩苷、黄芩素均能显著降低炎症时小鼠耳毛细血管通透性亢进及低气压所致小鼠试验性肺出血,对角叉菜胶性大鼠足肿有与阿司匹林相似的抗炎作用,对酵母、伤寒菌苗所致家兔发热有解热作用;黄芩还具有广谱的抗菌作用,对流感病毒、鼻病毒等有抑制作用。白芍为毛茛科植物芍药的干燥块茎,主要含有芍药苷及牡丹酚等,具有免疫调节作用,可提高小鼠腹腔巨噬细胞的吞噬百分率和吞噬指数;对T细胞功能呈机能与浓度依赖性双向调节作用,可促进特异性及非特异性调节细胞的诱导,具有明显的抗炎作用;还具有抗菌、抗病毒作用。甘草为豆科植物甘草的干燥根及根茎,含甘草甜素及多种黄酮类成份,具有多种生物活性。甘草酸类化合物通过对多种病毒的直接作用和诱生干扰素,增强天然杀伤细胞和巨噬细胞活性等活化宿主免疫功能的间接作用而发挥广谱的抗病毒作用,对艾滋病毒(HIV)、单纯性疱疹病毒、水痘-带状疱疹病毒(VZV)均有抑制作用,还具有诱导干扰素的作用,剌激细胞产生γ-干扰素,而γ-干扰素为国外报道具有抑制SARS病毒的作用,此外,甘草还有提高免疫功能、抗炎、解毒等作用。
“达原饮”传统用药剂型为水煎剂,为了更好的发挥达原饮的临床疗效,医药工作者对该经典方进行了大量的研究,主要是对其临床药理作用作了大量工作,对其质量研究报道较少;作为含七味中药的复方制剂,所含药味多,具有多组分、多靶标等特点,目前大部分中药复方制剂在含量控制上基本只有一个或两个指标性成分,质量控制单一,指标少,难以全面反映产品的质量状况。鉴于目前质量控制方法在全面控制产品质量上存在不足,同时达原饮中由于含有生物碱、黄酮类等理化性质差异较大成分,若针对每个指标单独建立方法测定,较繁琐。目前在达原饮全面质量控制方法的研究上未见报道,因此目前本领域亟需一种较全面的达原饮组合物的质量控制的方法。
技术实现要素:
因此,本发明要解决的技术问题在于克服现有技术中的中药复方制剂在含量控制上基本只有一个或两个指标性成分,质量控制单一,指标少,难以全面反映药味多,组分多、靶标多的达原饮组合物的质量状况的缺陷,从而提供达原饮组合物的质量控制方法。
为此,本发明提供如下技术方案:
达原饮组合物的质量控制方法包括采用高效液相色谱法建立达原饮组合物指纹图谱,色谱条件为:
色谱柱采用十八烷基硅烷键合硅胶色谱柱;
流速0.8ml/ml;柱温:25℃~35℃;
检测仪器采用紫外-可见分光检测器,指纹图谱的检测波长为215nm~240nm;
理论板数按参照物黄芩苷峰计算,应不低于3000;
参照物溶液为黄芩苷的甲醇溶液;
流动相以乙腈-0.1%磷酸溶液为系统按照如下洗脱顺序进行梯度洗脱:
指纹图谱流动相梯度程序
所述指纹图谱包括20个共有峰,各峰的相对保留时间分别为:
1号峰相对保留时间RRT为11.554,2号峰相对保留时间RRT为20.589,3号峰相对保留时间RRT为24.434,4号峰相对保留时间RRT为26.558,5号峰相对保留时间RRT为29.776,6号峰相对保留时间RRT为31.082,7号峰相对保留时间RRT为34.114,8号峰相对保留时间RRT为37.788,9号峰相对保留时间RRT为38.742,10号峰相对保留时间RRT为39.778,11号峰相对保留时间RRT为40.583,S峰相对保留时间RRT为50.099,13号峰相对保留时间RRT为53.925,14号峰相对保留时间RRT为54.927,15号峰相对保留时间RRT为57.315,16号峰相对保留时间RRT为61.652,17号峰相对保留时间RRT为68.702,18号峰相对保留时间RRT为72.735,19号峰相对保留时间RRT为84.317,20号峰相对保留时间RRT为86.291,其中,12号S峰是参照物的色谱峰。
所述达原饮组合物按如下两种方法制备:
(1)按重量份计称取如下饮片:槟榔6g,厚朴3g,草果仁1.5g,知母3g,白芍3g,黄芩3g,甘草1g,置于2L煎药砂锅中,加400ml水,浸泡30分钟,加盖,武火加热煮沸,文火保持微沸至药材药液为150-170ml,滤除药渣,放冷,即得第一达原饮组合物;
(2)按重量份计称取如下饮片:槟榔1200g,厚朴600g,草果仁300g,知母600g,白芍600g,黄芩600g,甘草300g;加8倍量的水,煎煮1.0h;滤过,得滤液;减压浓缩至相对密度1.10-1.30的稠膏,加入麦芽糊精,干燥;混匀,加硬脂酸镁,干压颗粒,制成1000g,即得第二达原饮组合物。
测定达原饮组合物指纹图谱时的供试品溶液与参照物溶液按如下方法制备:
(1)供试品溶液的制备:取第一达原饮组合物5ml,离心,取上清液,即得;或取第二达原饮组合物1.7g,置50ml容量瓶中,加水稀释,超声溶解,放冷,再加水定容,摇匀,离心,取上清液,即得;
(2)参照物溶液的制备:取黄芩苷对照品,加甲醇配制成80μg/ml的参照物溶液,即得。
进样量为10μl。
还包括用高效液相色谱法
测定达原饮组合物中槟榔碱含量,色谱条件为:
以强阳离子交换键合硅胶为填充剂;
流动相为乙腈-0.2%磷酸溶液为流动相(50:40)洗脱,2→1000,浓氨试液调节pH值至3.5-4.0;
流速0.8ml/ml~1.2ml/ml;柱温:25℃~35℃;
检测仪器采用紫外-可见分光检测器,槟榔碱含量测定的检测波长为215nm;
理论板数按对照品槟榔碱峰计算,应不低于3000;
对照品溶液为氢溴酸槟榔碱溶液。
测定达原饮组合物槟榔碱含量时的供试品溶液、对照品溶液按如下方法制得:
(1)供试品溶液的制备:取第一达原饮组合物5ml,离心,取上清液,即得;或取第二达原饮组合物,研细,取1g,精密称定,置于100ml容量瓶中,加50%乙腈超声溶解并定容至刻度,摇匀,离心,取上清液,即得;
(2)对照品溶液的制备:取氢溴酸槟榔碱,精密称定,加乙腈-0.2%磷酸溶液为流动相按50:50制成每1ml含40μg的溶液,即得,2→1000,浓氨试液调节pH值至约3.8,槟榔碱重量=氢溴酸槟榔碱重量/1.5214。
测定槟榔碱含量时的进样量为20μl~40μl。
还包括用高效液相色谱法
测定达原饮组合物中芒果苷、芍药苷、黄芩苷、甘草酸的含量,色谱条件为:
色谱柱采用十八烷基硅烷键合硅胶色谱柱;
流速0.9ml/min~1.1ml/min;柱温:25℃~35℃;
检测仪器采用二极管阵列检测器,四指标成分检测波长为230nm~254nm;
理论板数按对照品黄芩苷峰计算,应不低于5000;
对照品溶液为:芒果苷、芍药苷、黄芩苷、甘草酸铵的混合对照品溶液
流动相为乙腈-0.1%磷酸溶液为流动相按照如下洗脱顺序进行梯度洗脱:
流动相梯度程序
测定达原饮组合物中芒果苷、芍药苷、黄芩苷、甘草酸铵含量时供试品溶液和对照品溶液按如下方法制得:
(1)供试品溶液的制备:量取第一达原饮组合物5ml置于10ml的容量瓶中,加甲醇定容至刻度,摇匀,取适量离心,取上清液,即得;
或取第二达原饮组合物,研细,取0.5g,精密称定,置于100ml容量瓶中,加50%甲醇超声溶解并定容至刻度,摇匀,离心,取上清液,即得;
(2)对照品溶液的制备:取芒果苷对照品、芍药苷对照品、黄芩苷对照品、甘草酸铵对照品,称定,加70%甲醇分别制成每1ml含芒果苷20μg、芍药苷36μg、黄芩苷0.15mg、甘草酸铵20μl的溶液,即得,甘草酸重量=甘草酸铵重量/1.0207;
测定芒果苷、芍药苷、黄芩苷、甘草酸的含量时的进样量为10μl。
本发明提供的达原饮组合物的质量控制方法具有如下优点:
1、本发明提供的达原饮组合物的质量控制方法中的指纹图谱,全面反映出达原饮质量信息,从而能够达到更加全面、有效地控制达原饮组合物制剂产品质量的目的。
2、本发明提供的达原饮组合物的质量控制方法通过采用国家药典委员会提供的中药色谱指纹图谱相似度评价系统对所测指纹图谱的辨认,操作方便、快捷;而且,以此得出的相以度结果对制剂指纹图谱进行评价,结论较为客观、准确。
3、本发明提供的达原饮组合物的质量控制方法经过对供试品制备方法的考察及测定指纹图谱的仪器,色谱柱、流动相、检测波长等条件进行系统的优选,建立了指纹图谱测定条件并进行了方法学考察,在对多批本组合物指纹图谱检测结果的基础上,逐渐积累数据,提出了对照指纹图谱,作为本品指纹图谱标准,从而达到能够更全面、有效地控制制剂质量的目的。
4、本发明提供的达原饮组合物的质量控制方法采用国家药典委员会提供中药色谱指纹图谱相似度评价系统计算本组合物相似度,经多次试验研究,通过与计算相对保留时间及相对峰面积的方法相比较,所得出的评价结论基本一致,使用中药色谱指纹图谱相似度评价系统评价指纹图谱的相似度,操作方便、快捷,以其得出的相似度结果,对制剂指纹图谱进行评价,结论较为客观、准确。
5、本发明提供的达原饮组合物的质量控制方法采用指纹图谱较全面的表征组合物的质量信息,从整体上反映产品质量,同时针对处方七味药材,采用多波长切换技术建立其中五味(约占71%)的含量控制指标,针对在普通C18柱上难以保留的槟榔碱,采用强性阳离子交换树脂柱进行测定,同时改进供试品溶液的制备方法,以简单、便捷的方式快速制备供试品溶液,且测定结果准确、可靠;本发明考虑目前现有技术在控制复方中成药上往往只有一个或两个指标性成分,且缺乏指纹图谱整体表征产品质量信息,因此提出以指纹图谱结合多指标成分含量控制的方式全面的控制达原饮组合物质量。
6、本发明提供的达原饮组合物的质量控制方法提出采用指纹图谱,结合多波长切换技术测定达原饮组合物种多个指标成分。
7、本发明提供的达原饮组合物的质量控制方法可以为达原饮组合物提供一种较全面的质量控制方法。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1槟榔碱测量中对照品溶液色谱图;
图2槟榔碱测量中供试品溶液色谱图;
图3芒果苷、芍药苷、黄芩苷、甘草酸的测量中对照品溶液图
图4芒果苷、芍药苷、黄芩苷、甘草酸的测量中供试品溶液色谱图
图5指纹图谱测定中200~400nm扫描光谱3D图;
图6指纹图谱测定中的黄芩苷参照物溶液色谱图
图7指纹图谱测定中的对照指纹图谱
图8连续三批样品指纹图谱对比色谱图
具体实施方式
提供下述实施例是为了更好地进一步理解本发明,并不局限于所述最佳实施方式,不对本发明的内容和保护范围构成限制,任何人在本发明的启示下或是将本发明与其他现有技术的特征进行组合而得出的任何与本发明相同或相近似的产品,均落在本发明的保护范围之内。
实施例中未注明具体实验步骤或条件者,按照本领域内的文献所描述的常规实验步骤的操作或条件即可进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规试剂产品。
本发明试药及仪器:
试药:
氢溴酸槟榔碱(批号:111684-200401,中国药品生物制品检定研究院),
厚朴酚(批号:1100729-201513,中国药品生物制品检定研究院),
和厚朴酚(批号:110730-200402,中国药品生物制品检定研究院),
黄芩苷(批号:110715-201318,中国药品生物制品检定研究院),
芍药苷(批号:110736-201136,中国药品生物制品检定研究院),
芒果苷(批号:110607-200402,中国药品生物制品检定研究院),
甘草酸铵(批号:110731-201116,中国药品生物制品检定研究院);
槟榔(批号:20160210,产地:广西),
草果仁(批号:160127,产地:广西),
厚朴(批号:D1603007,产地:四川),
黄芩(批号:20160121,产地:山东),
白芍(批号:A160044001,产地:安徽),
知母(批号:A160059001,产地:河北),
甘草(批号:A160052001,产地:内蒙古)。
达原饮组合物①、达原饮组合物②(均自制,制备方法见实施例1),
乙睛,磷酸等,试验用水为超纯水;
仪器:Waters E2695高效液相色谱仪,Agilent 1260高效液相色谱仪
实施例1.达原饮组合物的制备
第一达原饮组合物:按重量份计称取如下饮片槟榔6g,厚朴3g,草果仁1.5g,知母3g,白芍3g,黄芩3g,甘草1g,置于2L煎药砂锅中,加400ml水,浸泡30分钟,加盖,砂锅置于电加热板上,武火(电压值220V)加热煮沸(约13分钟),文火(电压值约170V)保持微沸至药材药液约160ml(约40分钟),采用两层医用纱布滤除药渣,放冷,即得。
第二达原饮组合物:按重量份计称取如下饮片槟榔1200g,厚朴600g,草果仁300g,知母600g,白芍600g,黄芩600g,甘草300g;加8倍量的水,煎煮1.0h;滤过,得滤液;减压浓缩至相对密度为1.10-1.30优选1.20(60℃~70℃)的稠膏,加入适量麦芽糊精,干燥;混匀,加适量硬脂酸镁,干压颗粒,制成1000g,即得。第二达原饮组合物②采用聚酯镀铝/聚乙烯复合膜,包装规格为5.0g/袋。
实施例2.HPLC法测定达原饮组合物中槟榔碱含量
(1)色谱条件
以强阳离子交换键合硅胶为填充剂,色谱柱:Phenomenex Luna SCX,4.6*250mm,5um,以乙腈-磷酸溶液(2→1000,浓氨试液调节pH值至3.8)(50:50)为流动相;检测波长为215nm,流速1.0ml/min,柱温30℃。理论板数按槟榔碱峰计算应不低于3000。
(2)对照品溶液的制备
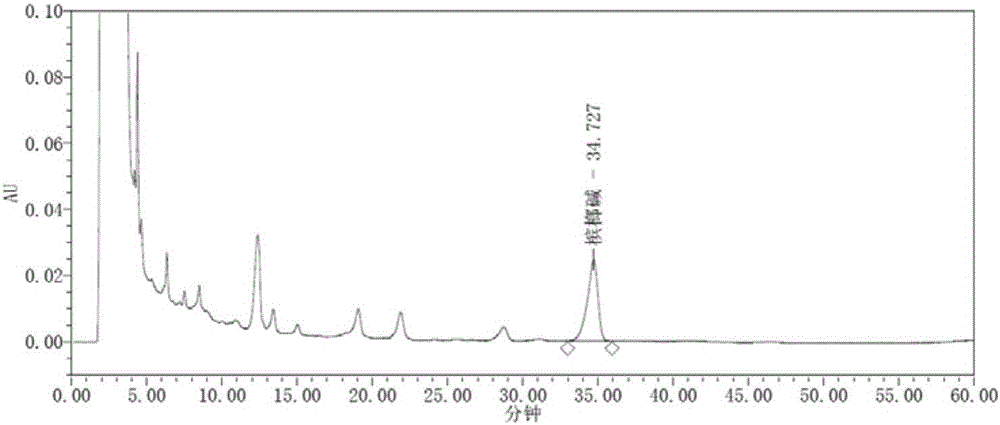
取氢溴酸槟榔碱适量,精密称定,加乙腈-0.2%磷酸溶液(2→1000,浓氨试液调节pH值至约3.8)为流动相(50:50)制成每1ml含40μg的溶液,即得。(槟榔碱重量=氢溴酸槟榔碱重量/1.5214)。见附图1。
(3)供试品溶液的制备
第一达原饮组合物供试品溶液制备:取第一达原饮组合物5ml,12000r/min离心10min,取上清液,即得。见附图2。
第二达原饮组合物供试品溶液制备:取第二达原饮组合物1.0g,精密称定,置100ml容量瓶中,加50%乙腈超声溶解并定容至刻度,摇匀,离心,取上清液,即得。
(4)测定法精密吸取对照品溶液与供试品溶液20μl~40μl,注入液相色谱仪,测定,即得。
(5)槟榔碱标准曲线的制备
精密移取一定量的对照品储备液,稀释,配制成浓度为0.81、4.06、6.50、8.12、9.75、20.30、40.61μg/ml的对照品溶液。分别进样20μl,按色谱条件进行测定,记录色谱图。以浓度(C)为横坐标,峰面积(A)为纵坐标,以浓度对平均峰面积进行回归分析,绘制标准曲线。线性回归方程为:A=141585C+16958,r=1,线性关系良好。
(6)方法学验证及样品测定该测定方法经过方法学验证,阴性样品对测定结果无干扰,样品在90h的稳定性良好,RSD值0.38%;重复性测定结果为RSD值为0.39%;平均加样回收率99.63%(n=9),RSD%为0.87%,表明方法准确度高,重复性良好。用本方法测定10份第一达原饮组合物与连续三批次第二达原饮组合物的槟榔碱含量,结果如下:
(7)第一达原饮组合物要求每份煎煮液、第二达原饮组合物要求槟榔碱含量每5g,两者均不得少于1.5mg。
实施例3.HPLC法测定达原饮组合物中芒果苷、芍药苷、黄芩苷、甘草酸的含量
(1)色谱条件:以十八烷基硅烷键合硅胶为填充剂,色谱柱:Agilent ZORBAX Eclipse Plus C18,4.6*250mm,5μm,以乙腈为流动相A,以0.2%磷酸溶液为流动相B,按下表中的规定进行梯度洗脱;流速0.8ml/min,柱温:30℃。理论板数按黄芩苷峰计算应不低于3000。
流动相梯度程序
检测波长表
(2)对照品溶液的制备
取芒果苷对照品、芍药苷对照品、黄芩苷对照品、甘草酸铵对照品适量,精密称定,加70%甲醇制成每1ml含芒果苷20μg、芍药苷36μg、黄芩苷0.15mg、甘草酸铵20μl的混合对照溶液,即得(甘草酸重量=甘草酸铵重量/1.0207)。见附图3。
(3)供试品溶液的制备
第一达原饮组合物供试品溶液制备:精密量取第一达原饮组合物5ml置于10ml的容量瓶中,加甲醇混匀并定容至刻度,取适量12000r/min离心10min,取上清液,即得第一达原饮组合物供试品溶液。见附图4。
第二达原饮组合物供试品溶液制备:取第二达原饮组合物0.5g,精密称定,置于100ml容量瓶中,加50%甲醇超声溶解并定容至刻度,摇匀,取适量12000r/min离心10min,取上清液,即得第二达原饮组合物供试品溶液。
(4)测定法 精密吸取对照品溶液与供试品溶液10μl,注入液相色谱仪,测定,即得。
(5)芒果苷、芍药苷、黄芩苷、甘草酸标准曲线的制备
精密移取一定量的混合对照品储备液,稀释,配制成含线性点1(芒果苷2.10、芍药苷1.73、黄芩苷23.97、甘草酸3.69μg/ml),线性点2(芒果苷10.52、芍药苷8.63、黄芩苷119.86、甘草酸18.47μg/ml),线性点3(芒果苷16.83、芍药苷13.81、黄芩苷191.78、甘草酸29.54μg/ml),线性点4(芒果苷21.04、芍药苷17.26、黄芩苷239.73、甘草酸36.93μg/ml),线性点5(芒果苷25.25、芍药苷20.72、黄芩苷287.67、甘草酸44.32μg/ml),线性点6(芒果苷52.60、芍药苷43.16、黄芩苷599.31、甘草酸92.33μg/ml),线性点7(芒果苷105.20、芍药苷86.32、黄芩苷1198.63、甘草酸184.65μg/ml)。分别进样10μl,按色谱条件进行测定,结果表明芒果苷线性回归方程为:A=2880112C+1777371,r=0.9999,线性关系良好;芍药苷线性回归方程为:A=2028361C-158727,r=1,线性关系良好;黄芩苷线性回归方程为:A=1214683C+6425261,r=1,线性关系良好;甘草酸线性回归方程为:A=1341078C+744191,r=0.9999,线性关系良好。其芒果苷浓度的线性范围为2.10~105.20μg·ml-1,芍药苷浓度的线性范围为3.69~184.65μg·ml-1,黄芩苷浓度的线性范围为23.97~1198.63μg·ml-1,甘草酸浓度的线性范围为1.73~86.32μg·ml-1。
(6)方法学考察及样品测定
该测定方法经过方法学验证,阴性样品对测定结果无干扰,样品在80h的稳定性良好,四个指标RSD值依次分别为:0.37%,0.76%,0.34%,0.28%;重复性测定结果四个指标RSD值分别为0.38%,0.47%,0.47%,0.56%;四个指标平均加样回收率分别为:芒果苷101.18%(n=9),RSD%为0.66%;芍药苷99.47%(n=9),RSD%为0.72%;黄芩苷100.02%(n=9),RSD%为0.69%;甘草酸103.29%(n=9),RSD%为0.63%,表明方法准确度高,重复性良好。用本方法测定10批次第一达原饮组合物与连续三批次第二达原饮组合物的四者含量,结果如下:
(7)第一达原饮组合物要求每份煎煮液、第二达原饮组合物要求每5g含量,两者芒果苷均不得少于4.4mg;芍药苷均不得少于13.3mg;黄芩苷均不得少于101.2mg;甘草酸均不得少于7.0mg。
实施例4.HPLC法建立达原饮组合物中指纹图谱的检测方法
(1)色谱条件及系统适用性以十八烷基硅烷键合硅胶为填充剂,色谱柱:waters symmetry C18 250mm*4.6mm,5μm;以乙腈为流动相A,0.1%磷酸溶液为流动相B;流速0.8ml/min,柱温30℃。理论板数按黄芩苷峰计算应不低于3000。
指纹图谱流动相梯度程序
检测波长表
(2)参照物溶液的制备 取黄芩苷对照品适量,加甲醇配制成250μg/ml的参照物溶液,即得。
(3)供试品溶液的制备
第一达原饮组合物供试品溶液:取第一达原饮组合物5ml,12000r/min离心10min,取上清液,即得第一达原饮组合物供试品溶液。
第二达原饮组合物供试品溶液:取第二达原饮组合物1.7g,精密称定,置于50ml量瓶中,加水超声溶解并定容至刻度,取适量12000r/min离心10min,取上清液,即得供试品溶液。
(4)测定法精密量取参照物溶液和供试品溶液10μl,注入高效液相色谱仪,测定,即得。
(5)达原饮组合物对照指纹图谱的确定及相似度分析
采用同一批次饮片,制备第一达原饮组合物10份,分别第一达原饮组合物供试品溶液,按指纹图谱测定法测定,记录图谱。通过分析,确定其共有特征峰为20个(见附图5-6),所述的20个共有特征峰的相对保留时间偏差RSD均小于2%,即:
1号峰平均保留时间RRT为11.554,RSD%为0.18%;
2号峰平均保留时间RRT为20.589,RSD%为0.03%;
3号峰平均保留时间RRT为24.434,RSD%为0.15%;
4号峰平均保留时间RRT为26.558,RSD%为0.14%;
5号峰平均保留时间RRT为29.776,RSD%为0.12%;
6号峰平均保留时间RRT为31.082,RSD%为0.10%;
7号峰平均保留时间RRT为34.114,RSD%为0.08%;
8号峰平均保留时间RRT为37.788,RSD%为0.11%;
9号峰平均保留时间RRT为38.742,RSD%为0.07%;
10号峰平均保留时间RRT为39.778,RSD%为0.07%;
11号峰平均保留时间RRT为40.583,RSD%为0.08%;
12(S)峰平均保留时间RRT为50.099,RSD%为0.07%;
13号峰平均保留时间RRT为53.925,RSD%为0.09%;
14号峰平均保留时间RRT为54.927,RSD%为0.11%;
15号峰平均保留时间RRT为57.315,RSD%为0.17%;
16号峰平均保留时间RRT为61.652,RSD%为0.18%;
17号峰平均保留时间RRT为68.702,RSD%为0.14%;
18号峰平均保留时间RRT为72.735,RSD%为0.05%;
19号峰平均保留时间RRT为84.317,RSD%为0.01%;
20号峰平均保留时间RRT为86.291,RSD%为0.01%;
运用中药色谱指纹图谱相似度评价软件(2012年版)处理图谱,以150914-1作为参照图谱,得到第一达原饮组合物对照指纹图谱。采用夹角余弦法计算样品相似度,结果表明,煎煮的10份第一达原饮组合物相似度在0.983~1.000之间,相似度较高。
10份第一达原饮组合物相似度评价结果
取第一达原饮组合物所对应的饮片,制备连续三批次第二达原饮组合物样品,按指纹图谱测定方法测定,记录色谱图(见附图7),以第一达原饮组合物生成的对照指纹图谱作为随行对照图谱,计算3批次第二达原饮组合物的相似度。结果见下表。
达原饮组合物②相似度计算结果
(6)按中药色谱指纹图谱相似度评价系统计算,以mark峰计,供试品指纹图谱与对照指纹图谱的相似度不得低于0.90。
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!