一种鉴定天麻产品的原料为新鲜天麻或干燥天麻的方法与流程
本发明涉及一种鉴定天麻产品的原料为新鲜天麻或干燥天麻的方法。
背景技术:
天麻是一味具有悠久历史的药食两用的名贵中药材,现在已经被开发为相关的中成药、食品饮料和保健酒等相关产品(Chau,C.F.,&Wu,S.H.(2006).The development of regulations of Chinese herbal medicines for both medicinal and food uses.Trends In Food Science&Technology,17(6),313-323;Yang,X.D.,Zhu,J.,Yang,R.,Liu,J.P.,Li,L.,&Zhang,H.B.(2007).Phenolic constituents from the rhizomes of Gastrodiaelata.Natural product research,21(2),180-186;Zhen,X.-J.,&Liu,J.-L.(2005).The technology research for Gastrodiaelata Bl.health protection beverage production.Food Science,26(9),653-655.)。由于天麻具有鲜、干两用的历史,所以在产品生产过程中既可投料新鲜的原材料,也可投放干燥样品。但是目前没有用于评价天麻产品新鲜或干燥原料投料的检测方法。为此,提供一种能够有效区分天麻产品中是否投有新鲜或干燥原料的方法具有重要意义。
技术实现要素:
本发明的目的是提供一种鉴定天麻产品的原料为新鲜天麻或干燥天麻的方法,本发明方法将两个特征性化学成分(式Ⅰ和式Ⅱ所示化合物)作为鉴定的标志物,能够有效区分天麻产品的原料投料形式—新鲜天麻或干燥天麻投料。
本发明首先提供式Ⅰ和式Ⅱ所示化合物:
式Ⅰ和式Ⅱ所示化合物可通过如下方法提取:将新鲜天麻研磨破碎后用水进行超声提取,离心收集上清液,分离提纯所述上清液即可。
式Ⅰ和式Ⅱ所示化合物还可由13#化合物依次经脱水缩合、水合及对羟基苯甲醇化修饰制备得到,各反应可在常规条件下进行;
式Ⅰ和/或式Ⅱ所示化合物可用作区分新鲜天麻或干燥天麻制备的天麻产品,即作为区分天麻产品的原料为新鲜天麻或干燥天麻的标志物,其中的天麻类型包括红天麻、黄天麻、乌天麻、绿天麻及相关的杂交品种。
本发明进一步提供的基于式Ⅰ和/或式Ⅱ所示化合物作为标志物的鉴定天麻产品的原料为新鲜天麻或干燥天麻的方法,当天麻产品中检测出式Ⅰ所示化合物和/或式Ⅱ所示化合物时,则所述天麻产品的原料为新鲜天麻,反之则为干燥天麻。
上述的方法中,所述天麻产品进行如下前处理:
当所述天麻产品为液态时,进行如下1)-3)中任一种处理:
1)所述天麻产品经离心收集上清液;
2)用水稀释所述天麻产品后经离心收集上清液;
3)所述天麻产品进行富集;
处理1)适用于含有合适浓度标志物的液态样品;
处理2)适用于含有较高浓度标志物的液态样品,稀释的程度为样品中检测不产生饱和信号为止;
处理3)适用于含有微量标志物的液态样品,所述富集的方式可为液液萃取、固相小柱萃取等;
当所述天麻产品为固态时,将所述天麻产品进行破碎得粉末样品,再进行如下1)或2)的处理:
1)用水对所述粉末样品进行超声提取,经离心收集上清液;
2)用水溶解所述粉末样品,经超声提取后进行富集处理;
处理1)适用于含有合适或较高浓度标志物的固态样品;
处理2)适用于含有较低浓度标志物的固态样品,所述富集的方式可为液液萃取、固相小柱萃取等。
上述的方法中,所述粉末样品的粒度可为10~200目;
处理1)中所述水的用量可为:3~100mL水/g所述粉末样品,具体可为4mL水/g所述粉末样品。
上述的方法中,处理1)和处理2)中所述超声提取的时间可为5~60min,具体可为30min;
处理1)中所述离心的速率可为10000~30000rpm,具体可为13000rpm,时间可为5~60min,具体可为10min。
上述的方法中,采用如下1)-7)中任一种方法进行检测:
1)超高效液相色谱-高分辨质谱联用(含UPLC-PDA-ESI-Q-TOF-MS)的方法;
2)液相色谱-质谱联用的方法;
3)紫外分光光度法;
4)红外光谱法;
5)近红外光谱法;
6)荧光光度法;
7)核磁共振波谱法。
上述的方法中,采用超高效液相色谱-高分辨质谱联用的方法时的检测条件如下:
柱温:20~50℃,具体可为45℃;
检测波长:269.5~270.5nm;
流速:0.2~0.8mL/min,具体可为0.5mL/min;
流动相:A相为体积浓度为0~0.15%的甲酸、乙酸、三氟乙酸、醋酸铵、氨水或甲酸铵的水溶液,具体可为0.1%的甲酸水溶液,作为水相,B相为体积浓度为0~0.15%的甲酸、乙酸、三氟乙酸、醋酸铵、氨水或甲酸铵的乙腈、乙醇或甲醇溶液,具体可 为0.1%的甲酸乙腈溶液,作为有机相;
具体洗脱梯度可为:0~4min,0~0.5%B相;4~6min,0.5%~2%B相;6~7min,2%~8%B相;7~12min,8%~12%B相;12~18min,12%~20%B相;18~24min,20%~40%B相;24~25min,40%~45%B相;25~31min,45%~70%B相;31~33min,70%~98%B相;33~35min,98%B相,均指的是体积百分含量。
上述的方法中,所述超高效液相色谱-高分辨质谱联用的方法中,高分辨质谱的检测条件如下:
检测模式:负离子模式;
检测范围:0~1500Da;
扫描时间:0.1~0.2s,具体可为0.2s;
检测时间:0~35min,具体可为26.5min;
高碰撞能量:30~50V;
毛细管电压:1.0~6.0kV,具体可为2.0kV;
锥孔电压:10~60V,具体可为40V;
源内温度:50~500℃,具体可为100℃;
脱气温度:300~800℃,具体可为450℃;
锥孔气流速度:20~100L/h,具体可为50L/h;
脱气流速:100~2000L/h,具体可为900L/h。
上述的方法中,可采用MassLynx V4.1软件(Waters公司,Milford,USA)进行数据处理。
上述的方法中,其余几种检测方法均可在常规的条件下进行,根据待测天麻样品中标志物的浓度高低来调整具体的检测条件,以能有效检测出标志物即可。
本发明提供的式Ⅰ和/或式Ⅱ所示化合物可作为区分天麻产品的原料为新鲜天麻或干燥天麻的标志物,能够对天麻产品的原料形式进行有效的鉴定,且检测方法简单易行,通过常规的前处理以及检测方法(如液相色谱-质谱联用的方法、紫外、红外、近红外、荧光、核磁等)即能实现标志物的检测。检测的天麻产品可以但不限于固态(如中成药)、液态样品(如饮料);检测天麻的类型包括红天麻、黄天麻、乌天麻、绿天麻及相关的杂交品种。
附图说明
图1为新鲜和干燥天麻的高分辨质谱总离子流图和在270nm下的液相图;
其中,IS表示芦丁;5#化合物表示巴利森苷G;13#化合物表示CA(0/p-HA/0);32#化合物表示式Ⅰ所示化合物,34#化合物表示式Ⅱ所示化合物。
图2为式Ⅰ和式Ⅱ所示化合物的加和离子和二级质谱图(分别为上图和下图)。
具体实施方式
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1、
1、标准品和试剂
标准品巴利森苷E购自成都普瑞法科技开发有限公司(成都,中国),纯度大于97%;芦丁购自中国食品药品检定研究院(北京,中国)。
质谱级的乙腈和甲酸分别购自Merck(Darmstadt,Germany)和Fisher Scientific(Geel,Belgium)公司。
去离子水由Thermo Scientific(Langenselbold,Germany)公司的Barnstead GenPure UV/UF制水系统制备。
2、材料与样品制备
新鲜白麻和母麻于2015年11月份采自云南彝良,经鉴定为兰科天麻属栽培品Gastrodiaelata Bl.f.glaucaS.Chow的块根。每一份样品立即清洗后,用吸水纸擦拭完样品表面,均匀分成两部分:一部分用于制备新鲜样品,另一部分用于制备对应的干燥样品。
新鲜样品的制备:
分别随机称取0.35g左右的已切成小块的新鲜天麻(包括白麻和母麻),置于2.0mL的EP管内,利用Scientz电动组织研磨仪(宁波,中国)在60Hz频率下破碎2min,得到粒度均一的破碎样品;破碎样品用4倍体积(约1.40mL)的水溶解后,超声提取30min,经13000rpm离心10min后,吸取上清液,保存于-80℃,备用。
干燥样品的制备:
取一部分新鲜样品置于75℃的干燥箱内,干燥至连续两次称重的差异不超过5mg为止;利用Scientz电动组织研磨仪(宁波,中国)在60Hz频率下破碎2min,破碎后得到粒度为80目的粉末,干燥粉末溶解在水中,超声提取30min后,13000rpm离心10min,吸取上清液,制备为100mg/mL的干燥样品的提取液,置于-80℃,备用。
3、UPLC-PDA-ESI-Q-TOF-MSE分析检测
上述制备的所有提取液(上清液)经解冻后,吸取100μL样品溶液与10μL的芦丁溶液(107μg/mL)混合,用于UPLC-PDA-ESI-Q-TOF-MSE定性和定量分析。
色谱条件如下:液相,ACQUITY I-CLASS UPLC;色谱柱,ACQUITYT3 column(1.8μm,2.1mm i.d.×100mm)(Waters,MA);预柱,VanGuardTM T3C18pre-column(1.8μm,2.1mm i.d.×10mm)(Waters,MA);流速,0.5mL/min;柱温:45℃;检测波长:270nm。流动相:A相,0.1%的甲酸水溶液(体积);B相,0.1%的甲酸乙腈溶液(体积)。色谱梯度:0~4min,0~0.5%B;4~6min,0.5~2%B;6~7min,2~8%B;7~12min,8~12%B;12~18min,12~20%B;18~24min,20~40%B;24~25min,40~45%B;25~31min,45~70%B;31~33min,70~98%B;33~35min,98%B。
质谱条件如下:仪器,高分辨质谱Waters Xevo G2-S Q-Tof-MS(Manchester,UK);检测模式,负离子模式;检测范围,100~1500Da;扫描时间,0.2s;检测时间,26.5min;高碰撞能量,30~50V;毛细管电压,2.0kV;锥孔电压,40V;源内温度,100℃;脱气温度,450℃;锥孔气流速度,50L/h;脱气流速,900L/h。数据处理软件为MassLynx V4.1软件(Waters公司,Milford,USA)。
4、结果
图1为新鲜和干燥天麻(包括白麻、母麻两种规格)的高分辨质谱总离子流图和在270nm下的液相图(平行进行三次)。
图1中,IS表示芦丁,5#化合物表示巴利森苷G,其结构式如下所示;13#化合物的结构式如下所示,32#化合物即为式Ⅰ所示化合物其结构式如下所示,34#化合物即为式Ⅱ所示化合物,其结构式如下所示:
由图1可以看出,新鲜和干燥的天麻样品具有显著差异,其中新鲜的白麻和母麻样品主要由巴利森苷G(5#)、CA(0/p-HA/0)(13#)、HdCA(CA(0/p-HA-p-HA/0)/p-HA-p-HA/0)(32#,式Ⅰ)和HdCA(CA(0/p-HA-p-HA-p-HA/0)/p-HA/0)(34#,式Ⅱ)组成,其中32#和34#化合物在三批干燥样品中均未检出。因此32#和34#化合物可作为检测天麻产品的原料为新鲜天麻或干燥天麻的标志物。
各化合物的分析和鉴定数据如下:
5#化合物的高分辨MS/MS数据为397.1122、191.0188、173.0085、129.0187和111.0083。经文献(Li,Z.,Wang,Y.,Ouyang,H.,Lu,Y.,Qiu,Y.,Feng,Y.,Jiang,H.,Zhou,X.,&Yang,S.(2015).A novel dereplication strategy for the identification of two new trace compounds in the extract of Gastrodiaelata using UHPLC/Q-TOF-MS/MS.Journal of Chromatography B,988,45-52.)和同分异构体巴利森苷E的标准品比对,可鉴定化合物为巴利森苷G。
13#化合物的高分辨MS/MS数据为595.1240([2M-H]-),489.0847、191.0193、173.0075和111.0075。经文献(Lai,C.J.S.,Zha,L.,Liu,D.H.,Kang,L.,Ma,X.,Zhan,Z.L.,Nan,T.G.,Yang,J.,Li,F.,Yuan,Y.,&Huang,L.Q.(2016).Global profiling and rapid matching of natural products using diagnostic product ion network and in silico analogue database:Gastrodiaelata as a case study.Journal of Chromatography A,1456,187-195.)比对,可鉴定为CA(0/p-HA/0)。
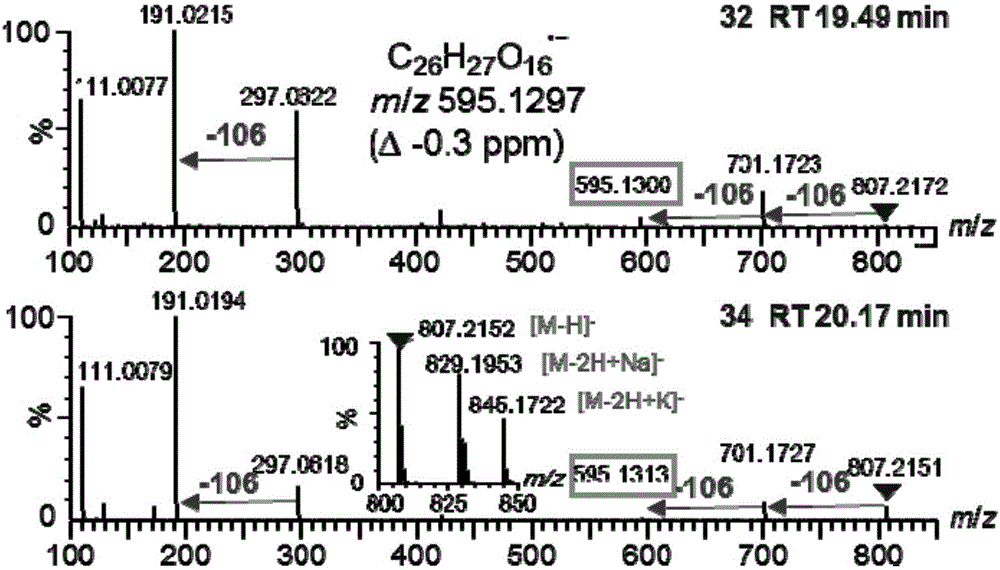
32#化合物和34#化合物为一对同分异构体,它们的高分辨质谱的母离子([M-H]-)和加和离子([M-2H+Na]-、[M-2H+K]-)数据表明该对同分异构体的分子式为C40H40O18。32#和34#化合物的二级质谱数据分别为701.1723、595.1300、297.0622、191.0215、1733.0072、111.0077;701.1727、595.1313、403.1015、297.0618、191.0194、173.0085、129.0184、111.0079。其中碎片离子m/z 297.06和m/z 595.13表明该对同分异构体具有一对含羟基对羟基苯甲醇柠檬酸骨架的化合物。由于柠檬酸酯类化合物sn-1/5键比sn-6更易断裂(Lai,C.J.S.,Zha,L.,Liu,D.H.,Kang,L.,Ma,X.,Zhan,Z.L.,Nan,T.G.,Yang,J.,Li,F.,Yuan,Y.,&Huang,L.Q.(2016).Global profiling and rapid matching of natural products using diagnostic product ion network and in silico analogue database:Gastrodiaelata as a case study.Journal of Chromatography A,1456,187-195.),因此高丰度的碎片离子m/z 297.06表明这对同分异构体的酸酐键在sn-1/5位。其中34#化合物的保留时间为20.17min大于32#化合物的保留时间19.49min,并且它们具有系列脱对羟基苯甲醇基(-106Da)的离子信号。通过分析生源合成途径,32#和34#化合物可由CA(0/p-HA/0)(13#)脱水缩合、水合及对羟基苯甲醇化修饰而成。所以32#和34#化合物分别鉴定为HdCA(CA(0/p-HA-p-HA/0)/p-HA-p-HA/0)(式Ⅰ)和HdCA(CA(0/p-HA-p-HA-p-HA/0)/p-HA/0)(式Ⅱ)。
- 还没有人留言评论。精彩留言会获得点赞!