一种含羟基的神经类固醇的检测分析方法与流程
本发明涉及分析化学领域,具体涉及一种含羟基的神经类固醇的检测分析方法,尤其是涉及一种首次合成并使用的d0/d3-3-n-甲基-2-羧基罗丹明6g(d0/d3-mcr6g)作为稳定同位素标记衍生试剂,衍生产物经磁分散固相萃取纯化和富集后,采用超高效液相色谱三重四极杆质谱检测的分析方法。
背景技术:
帕金森病(pd)是一种多发于中老年人的神经退行性疾病,随社会老龄化的加重,引起人们的广泛关注。有报道证明神经类固醇含量的变化与pd紧密相关并具有重要的研究意义,了解神经类固醇与pd之间的关系不仅有助于人们获得更高的生活质量,而且还有助于评估其神经保护作用以开发新的治疗方法。然而由于其含量低及生物样品中复杂的基质效应,以及神经类固醇在质谱负离子模式下有较弱的离子化效率,使其定量检测面临巨大的挑战。
现有技术中,采用多种不同的分析方法实现对神经类固醇的定量检测,如生物传感法、电化学检测法、液相色谱紫外检测法以及质谱检测等方法。文献《magneticsensingfilmbasedonfe3o4@au-gshmolecularlyimprintedpolymersfortheelectrochemicaldetectionofestradiol》(biosensorsandbioelectronics79(2016)180–186)采用电化学检测的方法实现对奶粉中雌二醇的定量检测。文献《anewone-stepantigenheterologoushomogeneousfluorescenceimmunoassayforprogesteronedetectioninserum》(talanta134(2015)508–513)采用均相免疫分析方法实现对人血清中孕酮的含量测定。然而以上两种方法在应用到实际生物样品中时,由于神经类固醇在生物样品中浓度低,且存在内源性干扰等,方法的灵敏度和准确度不足。文献《lc–ms/mssimultaneousanalysisofallopregnanolone,epiallopregnanolone,pregnanolone,dehydroepiandrosteroneanddehydroepiandrosterone3-sulfateinhumanplasma》(bioanalysis(2017)9(6),527–539)采用2-肼基吡啶作为衍生试剂对人体血浆中的神经类固醇定量检测,相比而言使用该方法对多种神经类固醇的检测,在色谱分离度和质谱选择性、灵敏度方面有所提高,但该方法直接检测存在潜在的色谱分离干扰、质谱检测离子化效率较低的缺点。专利(cn104807921a)先利用高效液相色谱将人血清中的10种类固醇激素分离,再利用质谱同位素内标的方法定量检测,该方法一定程度上提高了检测的灵敏度、准确度。但是并非所有的生物代谢物均可找到商品化的同位素标准品,且该类标准品价格较为昂贵。因此,设计合成合适的同位素标记衍生试剂,开发一种对实现多种神经类固醇同时高灵敏、高准确性的检测具有重要意义。
技术实现要素:
针对现有技术中存在的如何实现简单快速、准确且灵敏的对多种神经类固醇的同时定量检测的问题,本发明的目的在于提供一种含羟基的神经类固醇的检测分析方法,该方法采用首次合成的d0/d3-3-n-甲基-2-羧基-罗丹明6g(d0/d3-mcr6g)作为稳定同位素标记衍生试剂,对实际样品的检测结合了磁分散固相萃取技术进行纯化和富集,减小了基质干扰效应,提高了检测灵敏度和准确度。
本发明为了实现上述目的所采用的技术方案为:
本发明提供了一种含羟基的神经类固醇的检测分析方法,该方法以d0/d3-3-n-甲基-2-羧基罗丹明6g(d0/d3-mcr6g)作为同位素标记衍生试剂,用轻/重d0/d3-mcr6g分别标记衍生实际样品和标准品,以d3-mcr6g标记的标准品作为内标,得到的轻/重标记衍生化产物混合,使用磁分散固相萃取技术纯化和富集,经滤膜过滤后利用超高效液相色谱三重四极杆串联质谱系统进行分析检测。
本发明所提供的检测分析方法具体包括以下步骤:
a.稳定同位素标记衍生:将20-100μl混合标准品或待测实际样品分别置于离心管中,将d0-mcr6g乙腈溶液加入待测实际样品中,并将d3-mcr6g乙腈溶液快速注入标准溶液中,然后分别加入50μl2-氯-1-甲基碘代吡啶(cmpi)和50μl4-二甲氨基吡啶(dmap)乙腈溶液,密封两个离心管,超声波震荡30秒,在微波反应器中进行衍生反应,将d0-mcr6g标记的待测实际样品和d3-mcr6g标记的标准品1:1(v/v)混合,摇匀得衍生产物溶液;
b.磁分散固相萃取及分析:取4-12mg四氧化三铁修饰的氧化石墨烯(fe3o4/go)纳米材料通过超声分散到上述衍生产物溶液中;然后涡旋振荡2-10min后,采用外加磁铁的方式实现混合物的分离;将沉淀物中加入0.2-0.8ml解吸溶液,涡旋振荡6min解吸,然后用外部磁场分离,收集上层清液经滤膜过滤后利用超高效液相色谱三重四极杆串联质谱系统进行分析检测。
进一步的,所述2-氯-1-甲基碘代吡啶乙腈溶液的浓度为4–15wt%;所述4-二甲氨基吡啶乙腈溶液的浓度为1–10wt%;所述d0/d3-mcr6g乙腈溶液的摩尔浓度均为1×10-4mol/l;所述微波反应器的功率为750w,所述衍生反应为在30-85℃条件下反应5-35min;优化的衍生反应的温度为70℃,反应的时间为15min;所述d0-mcr6g乙腈溶液或d3-mcr6g乙腈溶液相比于或待测实际样品或混合标准品过量的倍数为10-300倍。
本发明所使用的衍生试剂d0/d3-3-n-甲基-2-羧基罗丹明6g(d0/d3-mcr6g)采用以下方法制备而成:将1g罗丹明6g溶于30mldmf中,室温下搅拌5分钟后,缓慢加入0.217gnah反应3分钟,然后在3-5分钟内加入330μlch3i或cd3i,室温反应150分钟后,加入150ml水终止反应,用150ml氯仿萃取混合物两次,合并有机层并减压蒸馏得粗产物,经甲醇/氯仿(1:1,v/v)重结晶,得到紫红色固体产物d0-mcr6g或d3-mcr6g。
进一步的,所述的四氧化三铁修饰的氧化石墨烯(fe3o4/go)纳米材料采用以下方法制备得到:将0.5gfe3o4分散在1mhno3中,然后0.1g氧化石墨烯超声处理30min制备得到1mg/ml的氧化石墨烯水溶液分散体,将上述得到的fe3o4溶液添加到go分散液中,机械搅拌30min后,经离心并施加外部磁铁从深黑色溶液分离出fe3o4/go固体,将该固体用高纯水洗涤5次,并在真空烘箱中60℃下干燥得到所述磁性材料。
进一步的,所述解吸溶液为含0.1%的甲酸的乙醇、乙腈、丙酮或甲醇;所述解吸溶液优化的为含0.1%甲酸的甲醇。
本发明所检测的神经类固醇为含有活性羟基的如下化合物:α-雌二醇、四氢孕酮、去氢表雄酮、睾酮、孕烯醇酮、17α-羟基孕烯醇酮、17α-羟孕酮。
进一步的,所述超高效液相色谱三重四极杆串联质谱系统中色谱分离使用安捷伦sbc18色谱柱:2.1mm×50mm,1.8µm,进样体积为2µl,柱温30℃,采用线性梯度洗脱法。
上述线性梯度洗脱法,时间为10min,流速为0.2ml/min,流动相a为5%乙腈水溶液含有0.1%甲酸,流动相b为乙腈含有0.1%甲酸,0min流动相组成为80%a+20%b,5min流动相组成为25%a+75%b,7min流动相组成为5%a+95%b,10min流动相组成为0%a+100%b。
进一步的,所述质谱的条件为:干燥气温度300℃,流速10l/min,喷雾器气压40psi,鞘气温度280℃,流速11l/min,毛细管电压3.5kv。
本发明所述nah采用煤油作为载体;所述nah在煤油中的质量百分含量为60%。
本发明提供了一种同时检测多种神经类固醇的分析方法,d0-mcr6g用于衍生实际样品中神经类固醇,同时d3-mcr6g衍生神经类固醇标准品作为内标,得到的衍生化产物1:1(v/v)混合后,经磁分散固相萃取纯化和富集,过滤膜后利用超高效液相色谱三重四极杆串联质谱系统进行分析检测。
本发明流动相中各分数均指体积分数。
所述的神经类固醇样品为帕金森病大鼠的脑微透析液、血清、尿液、脑组织的实际样品。
本发明所使用的超高效液相色谱三重四极杆串联质谱系统由安捷伦1290系列超高效液相色谱,配备安捷伦6460三重四极杆串联质谱系统组成。
所述的神经类固醇化学结构和代谢关系如下:
本发明利用同位素标记衍生技术结合磁分散固相萃取联合超高效液相色谱三重四极杆质谱法,实现对多种神经类固醇同时检测,提高了色谱分离能力和质谱离子化效率,进而提高检测灵敏度和准确度。首次合成并使用的d0/d3-3-n-甲基-2-羧基罗丹明6g(d0/d3-mcr6g)作为同位素标记衍生试剂,d0-mcr6g衍生实际样品,d3-mcr6g衍生标准品作为内标,二者1:1(v/v)混合后进样分析,由于轻重标记的产物在色谱图上近乎相同时间出峰,而在质谱信号(母离子及二级子离子)中的差别即为轻重标衍生试剂的m/z的差别,用重标标记的衍生物做内标可消除仪器波动影响及最小化基质效应,实现对实际样品中目标物更准确的定量分析检测。同时该衍生试剂自身带有正电荷,可显著提高质谱的离子化效率提高质谱检测。本发明为检测多种神经类固醇提供了一种灵敏快速、可靠的技术手段。神经类固醇与3-n-甲基-2-羧基-罗丹明6g衍生试剂的衍生化反应过程如下所示:
本发明的优点及有益效果:
(1)本发明设计合成的自身带有一分子正电荷的d0/d3-3-n-甲基-2-羧基罗丹明6g(d0/d3-mcr6g)做衍生试剂,可在液质联用多反应检测模式中提高色谱的分离能力和质谱的离子化效率,进而提高检测的灵敏度,而且衍生反应快速、产物稳定。
(2)采用合成的轻重标衍生试剂分别衍生实际样品和标准品,重标标记的衍生试剂衍生标准品作为内标,在色谱分离中二者近乎同时出峰,但在质谱结果中根据m/z值的不同实现相对定量,可以消除质谱波动的影响及最小化基质效应,进而提高分析物检测的准确度。
(3)结合磁分散固相萃取技术,使用四氧化三铁修饰的氧化石墨烯展现了较好的吸附能力,同时使用外加磁铁的方法分离,该方法经济、方便有效。
(4)本发明采用稳定同位素衍生结合磁分散固相萃取技术,联合超高效液相色谱三重四极杆质谱检测的分析方法,具有简单快速,高灵敏度和准确度的优点,同时对微透析液、血清等实际样品中的神经类固醇的检测展现了较好的适用性。
附图说明
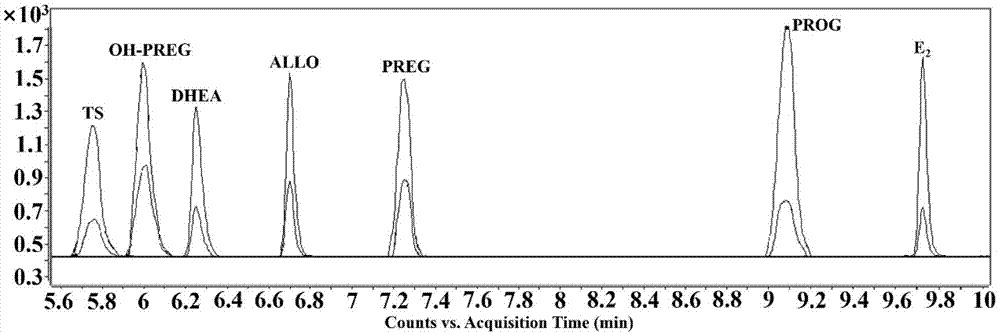
图1为实施例1中7种神经类固醇标准品衍生物质谱检测分离图。
图2为实施例1衍生物质谱裂解机理示意图:(a)d0-mcr6g-四氢孕酮衍生物;(b)d3-mcr6g-四氢孕酮衍生物。
图3为实施例2中帕金森病大鼠脑微透析液的质谱检测分离图四氢孕酮。
具体实施方式
下面通过实施例对本发明进行进一步的阐述,下述说明仅为了解释本发明,并不对其内容进行限定。
本发明所使用的甲醇、丙酮、乙腈等化学试剂为色谱纯,水为高纯水。
本发明中衍生化萃取物经滤膜(0.45μm)过滤后利用超高效液相色谱三重四极杆串联质谱系统进行分析检测。
实施例1
神经类固醇的色谱分离和质谱定性定量分析方法:
神经类固醇的标准品(购自sigma试剂公司)用乙腈做溶剂配制,在10-2500pg/ml范围内对每一种神经类固醇配制7个不同浓度d0-mcr6g标记的标准溶液(10pg/ml,100pg/ml,500pg/ml,1000pg/ml,1500pg/ml,2000pg/ml,2500pg/ml),其中以d3-mcr6g标记的标准品(200pg/ml)作为固定内标。具体的衍生过程如下:取100μl不同浓度的混合标准品1份,150μld0-mcr6g或d3-mcr6g乙腈溶液,50μl2-氯-1-甲基碘代吡啶(cmpi5%)和50μl4-二甲氨基吡啶(dmap10%)乙腈溶液,依次加入到具有圆锥形底部的1.5ml离心管中。密封两个离心管,超声波震荡30秒以充分混合。将管密封并在70℃在微波反应器(750w)中辐照15min。将7个不同浓度水平的d0-mcr6g混标衍生物和d3-mcr6g标记的固定混标衍生物分别1:1(v/v)混合,摇匀。磁分散固相萃取过程如下:混合后的溶液加入到8mgfe3o4/go纳米复合材料经超声得到的均匀溶液中,涡旋摇晃6min达到吸附平衡后,混合物通过外加磁铁的方法分离,倾去上清液,然后向剩余的沉淀中加入0.4ml含0.1%甲酸的甲醇洗脱,涡旋震荡6min实现解吸,然后用外部磁铁分离,收集到的上清液经氮气吹干,200μl甲醇复溶,经有机滤膜过滤后进样2µl,进行超高效液相色谱三重四极杆质谱法检测分析。依据d0-/d3-mcr6g衍生物的峰面积比值(d0-mcr6g混标衍生物/d3-mcr6g固定内标)与d0-mcr6g标记的标准品浓度进行线性回归,制作校准曲线,按照上述分析过程对实际样品进行测定的峰面积带入线性方程计算实际样品神经类固醇的含量。
流动相a为5%乙腈水溶液含有0.1%甲酸,流动相b为乙腈含有0.1%甲酸,按照上述部分的线性梯度洗脱程序能得到较好的分离度,图1为7种神经类固醇及内标物的衍生物质谱检测分离图,7种分析物之间分离度良好。质谱分析结果表明,各种神经类固醇经d0/d3-mcr6g衍生后得到的衍生产物在多反应监测模式(mrm)下产生一对相同的子离子分别为m/z429.2和m/z432.2。以四氢孕酮衍生物为例,图2为四氢孕酮衍生物质谱裂解机理示意图,d0-mcr6g标记的母离子为m/z729.4,在mrm检测时产生特异子离子信号m/z429.2,d3-mcr6g标记的母离子为m/z732.4,在mrm检测时产生特异子离子信号m/z432.2。对神经类固醇mrm检测模式的参数进行了优化,分析物的检出限、定量限、衍生效率、最优化碎裂电压(v)和对应的二级信号以及线性方程的相关系数等参数见表1。
表1
实施例2
大鼠分组、给药及微透析取样检测:
将16只成年雄性sprague-dawley大鼠(体重220±30g)随机分为两组安置在标准条件下,自由饮水进食。帕金森病造模过程及行为学评价与文献(pharmacologybiochemistryandbehavior2011,98:286-291)保持一致。在脑微透析探针植入手术前用20%尿烷(1.2g/kg,i.p.)麻醉,并在整个实验期间保持麻醉。实验动物分成两组(n=8):a组,正常大鼠;b组,pd组大鼠,按照活体微透析取样的基本流程进行操作,微透析泵流速2.0μl/min,两组每只大鼠在前120min进行平衡,之后按每10min收集20μl微透析液的速度获取微透析液样品,每份样品经氮吹干后用等量乙腈溶解,待后续测定。
之后将200μld0-mcr6g乙腈溶液加入所取的微透析样品中,同时200μld3-mcr6g乙腈溶液加入到神经类固醇混标样品中。然后加入50μlcmpi(4%)和50μldmap(8%)乙腈溶液。密封两个离心管,超声波震荡30秒以充分混合。将管密封并在65℃微波反应器(700w)中辐照25min。实际样品和标准品的衍生化产物溶液1:1(v/v)混合摇匀,加入到10mgfe3o4/go纳米复合材料经超声得到的均匀溶液中,涡旋摇晃8min达到吸附平衡后,混合物通过外加磁铁的方法分离,倾去上清液,然后向剩余的沉淀中加入0.3ml含0.1%甲酸的乙腈,涡旋震荡8min实现解吸,然后用外部磁铁分离,收集到的上清液经氮气吹干,200μl甲醇复溶,经有机滤膜过滤后进样2µl,进行超高效液相色谱三重四极杆质谱法检测分析。脑微透析液中含各种神经类固醇的含量根据实施例1建立的定量方程计算得出。
实施例3
帕金森病大鼠血清样品的检测:
按照实施例2中的造模,对帕金森病大鼠的颈下取血,取得的血清样品经乙腈:水=9:1的方式提取并脱除蛋白,在4℃,15000r,离心15min,上清液过滤膜后,取100μl氮吹干后用等体积乙腈溶解,向其中加入100μld0-mcr6g乙腈溶液加入所取的提取后的血清样品中,同时100μld3-mcr6g乙腈溶液加入到神经类固醇混标样品中。然后再同时都加入50μlcmpi(7%)和50μldmap(9%)乙腈溶液。密封两个离心管,超声波震荡30秒以充分混合。将管密封并在55℃在微波反应器(600w)中辐照30min。实际样品和标准品的衍生化产物溶液1:1(v/v)混合后,加入到7mgfe3o4/go纳米复合材料经超声得到到均匀的溶液中,涡旋摇晃10min达到吸附平衡后,混合物通过外加磁铁的方法分离,倾去上清液,然后向剩余的沉淀中加入0.3ml含0.1%甲酸的乙醇,涡旋震荡10min实现解吸,然后用外部磁铁分离,收集到的上清液经氮气吹干,200μl甲醇复溶,经有机滤膜过滤后进样2µl,进行超高效液相色谱三重四极杆质谱法检测分析。血清中含各种神经类固醇的含量根据实施例1建立的定量方程计算得出。
实施例4
帕金森病大鼠尿液样品的检测:
将上述实施例2中不同阶段的大鼠分别搜集尿样,各取50µl,经4℃,15000r,离心15min后,取上清液氮吹干后用等量乙腈溶解,加入250μld0-mcr6g乙腈溶液,同时250μld3-mcr6g乙腈溶液加入到神经类固醇混标样品中。然后再同时都加入50μlcmpi(5%)和50μldmap(12%)乙腈溶液。密封两个离心管,超声波震荡30秒以充分混合。将管密封并在45℃在微波反应器(460w)中辐照50min。实际样品和标准品的衍生化产物溶液1:1(v/v)混合后,加入到6mgfe3o4/go纳米复合材料经超声得到的均匀溶液中,涡旋摇晃15min达到吸附平衡后,混合物通过外加磁铁的方法分离,倾去上清液,然后向剩余的沉淀中加入0.5ml含0.1%甲酸的丙酮,涡旋震荡15min实现解吸,然后用外部磁铁分离,收集到的上清液经氮气吹干,200μl甲醇复溶,经有机滤膜过滤后进样2µl,进行超高效液相色谱三重四极杆质谱法检测分析。尿液中含各种神经类固醇的含量根据实施例1建立的定量方程计算得出。
实施例5
帕金森病大鼠脑组织样品的检测:
按照生物学惯用的获取脑组织的方法获得大鼠端脑组织,经负80℃液氮冷冻后,准确称取0.05g大鼠脑组织样品于5ml离心管中,加入乙腈进行常规组织匀浆10min,涡旋30s超声3min,经4℃,15000r,离心15min后,取上清液氮吹干后加入等量乙腈溶解,加入200μld0-mcr6g乙腈溶液,同时200μld3-mcr6g乙腈溶液加入到神经类固醇混标样品中。然后再同时都加入50μlcmpi(6%)和50μldmap(12%)乙腈溶液。密封两个离心管,超声波震荡30秒以充分混合。将管密封并在60℃在微波反应器(650w)中辐照25min。实际样品和标准品的衍生化产物溶液1:1(v/v)混合后,加入到12mgfe3o4/go纳米复合材料经超声得到到均匀的溶液中,涡旋摇晃5min达到吸附平衡后,混合物通过外加磁铁的方法分离,倾去上清液,然后向剩余的沉淀中加入0.3ml含0.1%甲酸的甲醇,涡旋震荡5min实现解吸,然后用外部磁铁分离,收集到的上清液经氮气吹干,200μl甲醇复溶,经有机滤膜过滤后进样2µl,进行超高效液相色谱三重四极杆质谱法检测分析。脑组织样品中含各种神经类固醇的含量根据实施例1建立的定量方程计算得出。
将实施例1-5进行超高效液相色谱三重四极杆质谱法检测分析,使用安捷伦sbc18色谱柱:2.1mm×50mm,1.8µm,进样体积为2µl,柱温30℃,采用线性梯度洗脱法,时间为10min,流速为0.2ml/min,流动相a为5%乙腈水溶液含有0.1%甲酸,流动相b为乙腈含有0.1%甲酸,0min流动相组成为80%a+20%b,5min流动相组成为25%a+75%b,7min时流动相组成为5%a+95%b,10min流动相组成为0%a+100%b;所述质谱的条件为:干燥气温度300℃,流速10l/min,喷雾器气压40psi,鞘气温度280℃,流速11l/min,毛细管电压3.5kv。检测到帕金森病大鼠不同样品中神经类固醇的含量(n=3)如表2所示。
表2
-:未检测到。
对比例1
该对比例过程与实施例2相同,不同在于衍生化反应过程中,采用商品化丹磺酰氯作为衍生试剂进行对比。
对比例2
该对比例过程与实施例2相同,不同在于衍生化反应过程中,采用n-甲基-n-三甲基硅基三氟乙酰胺作为衍生试剂。
下表3为实施例2同对比例1和2的实验结果对比。
表3
表3结果表明,本发明利用同位素标记衍生技术和磁分散固相萃取技术相结合,具有高灵敏度、高准确度、简单快速方便的特点。本发明的检出限比对比例低166-3333倍左右。
本分析方法中对多种神经类固醇检测的线性范围是10-2500pg/ml,在最优检测条件下分析物神经类固醇的线性相关系数r2>0.99,检出限分布于0.06-0.12pg/ml之间,定量限分布于0.3-0.4pg/ml之间。表1-2结果表明,所建立的分析方法能够很好地应用于不同的实际生物样品中神经类固醇含量测定。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受实施例的限制,其它任何未背离本发明的精神实质与原理下所做的改变、修饰、组合、替代、简化均应为等效替换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!