红参药材指纹图谱及其与制剂成分关联性的构建方法与流程
本发明涉及中药质检技术领域,尤其是涉及红参药材指纹图谱及其与制剂成分关联性的构建方法。
背景技术:
中药指纹图谱是指某种中药材及其制剂中所共有的、具有特征性的某类或数类成分的色谱或光谱的图谱,指纹图谱的建立对于有效控制药材及其制剂的质量具有十分重要的意义。
红参为五加科植物人参panaxginsengc.a.mey.的栽培品经蒸制后的干燥根和根茎,有大补元气,复脉固脱,益气摄血之效。现阶段红参在《中国药典》中只是对人参皂苷含量进行控制,未对药材建立hplc指纹图谱,不能达到对药材及其制剂全面质量评价的目的。指纹图谱的建立对于有效控制红参及其制剂的质量具有十分重要的意义。
有鉴于此,特提出本发明。
技术实现要素:
本发明的第一目的在于提供红参药材指纹图谱的构建方法,该构建方法能够准确测定红参药材中的主要成分,为红参药材的质量评价提供了可靠的指纹图谱,对于有效控制红参药材的质量具有十分重要的意义。
本发明的第二目的在于红参药材与其制剂成分关联性的构建方法,该构建方法能将红参药材与制剂之间相关成分关联,能对药材与制剂的质量相关进行评价。
为了实现以上目的,本发明提供了以下技术方案:
红参药材指纹图谱的构建方法,包括下列步骤:
以80~90%的乙醇为溶剂,对红参药材浸提,得到药材样品溶液;
采用高效液相色谱仪检测所述药材样品溶液,得到指纹图谱;
所述检测的色谱条件为:
色谱柱:反相色谱柱,
流速:0.5~1.5ml/min,
检测波长200-210nm,
柱温:25~35℃,
进样量:8~15μl,
流动相:乙腈和水以0:100~100:0的体积比梯度洗脱,
所述检测时所用的对照品为人参皂苷rg1、re和rb1。
本发明的以上方法通过特定的样品处理以及色谱条件对红参药材中的多种活性成分有效分离,并定量检测。
本发明的以上方法至少能检测出红参药材中的22个特征峰,并且22个特征峰之间具有明显的分离度,本发明指认了其中9个特征峰的化合物结构,分别为:五-羟甲基糠醛、核苷、人参皂苷rg1、人参皂苷re、人参皂苷rf、人参皂苷rb1、人参皂苷rc、人参皂苷rb2、人参皂苷rd。
本发明中,浸提红参的乙醇浓度可选80~90%范围内的任意值,例如80%、81%、82%、83%、84%、85%、86%、87%、88%、89%等。
本发明可采用任意类型的反相色谱柱,例如rp18(4.6mm×250mm;5.0μm)。
本发明可选用0.5~1.5ml/min范围内的任意流速,例如0.5ml/min、0.6ml/min、0.7ml/min、0.8ml/min、0.9ml/min、1.1ml/min、1.2ml/min、1.3ml/min等。
本发明可选用200-210nm范围内的检测波长,例如200nm、203nm、203nm、205nm、207nm、210nm等。
本发明可选用25~35℃范围内的柱温,例如25℃、26℃、27℃、28℃、29℃、30℃、33℃、35℃等。
本发明可选用8~15μl范围内的进样量,例如8μl、9μl、10μl、11μl、12μl、15μl等。
本发明为梯度洗脱,起始体积比为乙腈/水=0:100,终点体积比为乙腈/水=0:100。
在以上基础上,更优选的色谱条件如下。
优选地,所述溶剂为85~90%的乙醇。
优选地,所述检测的色谱条件为:
流速:1~1.5ml/min,优选1~1.3ml/min,
检测波长203-210nm,优选203-208nm,
柱温:25~35℃,优选30~35℃,
进样量:8~15μl,优选10~15μl,
流动相:梯度洗脱按照表1进行:
表1梯度洗脱
本发明的浸提手段不受限制,可以超声或者回流。优选回流,成本低,易操作。
优选地,所述浸提为回流提取4小时以上。
优选地,所述回流提取的料液比为1g:5~15ml,优选1g:10~15ml。
优选地,所述检测时配制的对照品溶液所用的溶剂为15~25%乙腈,优选20~25%乙腈。
优选地,所述对照品溶液的配制方法为:
分别将人参皂苷rg1、re和rb1配成0.08~0.15mg/ml、0.05~0.1mg/ml和0.15~0.25mg/ml的溶液。
在以上基础上,本发明还建立了红参药材与其制剂成分关联性的构建方法,包括下列步骤:
a:以80~90%的乙醇为溶剂,对红参药材浸提,得到药材样品溶液;
采用高效液相色谱仪检测所述药材样品溶液,得到药材的指纹图谱;
所述检测的色谱条件为:
色谱柱:反相色谱柱,
流速:0.5~1.5ml/min,
检测波长200-210nm,
柱温:25~35℃,
进样量:8~15μl,
流动相:乙腈和水以0:100~100:0的体积比梯度洗脱,
所述检测时所用的对照品为人参皂苷rg1、re和rb1。
b:按照所述红参制剂生产工艺过程制得制剂样品溶液,采用与所述步骤a相同的色谱条件检测所述样品溶液,得到制剂的指纹图谱;
以所述药材的指纹图谱中的人参皂苷rb1特征峰为参照峰,计算出所述药材的指纹图谱中其他特征峰的相对保留时间;
以所述制剂的指纹图谱中的人参皂苷rb1特征峰为参照峰,计算出所述制剂的指纹图谱中其他特征峰的相对保留时间;
对比所述药材的指纹图谱中特征峰的相对保留时间和所述制剂的指纹图谱中特征峰的相对保留时间,近似度为0.9以上的则为相同成分。
优选地,所述制剂为生脉注射液或回阳复脉注射液。
实际应用时,本发明的方法并不仅仅适用于以上两种注射液。
优选地,所述近似度为0.95以上。
综上,与现有技术相比,本发明达到了以下技术效果:
(1)能够准确测定红参药材中的主要成分,为红参药材的质量评价提供了可靠的指纹图谱,对于有效控制红参药材的质量具有十分重要的意义;
(2)提供了评价原料与制剂之间相关成分关联性的方法。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
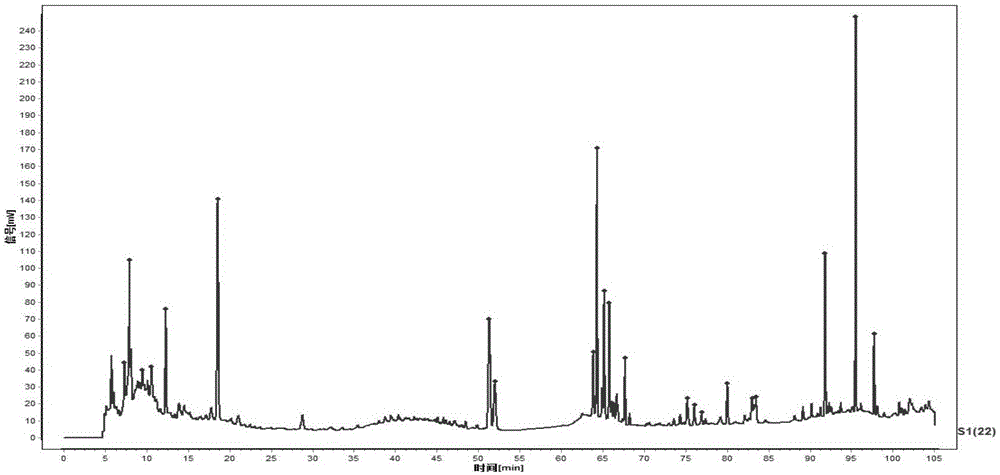
图1为本发明实施例提供的红参药材指纹图谱;
图2为本发明实施例提供的生脉注射液指纹图谱。
具体实施方式
下面将结合附图和具体实施方式对本发明的技术方案进行清楚、完整地描述,但是本领域技术人员将会理解,下列所描述的实施例是本发明一部分实施例,而不是全部的实施例,仅用于说明本发明,而不应视为限制本发明的范围。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
实施例1
(1)供试品溶液的制备:
制备红参药材提取液,精密称定2g红参,加入20ml90%乙醇,回流4小时,过滤,收集续滤液作为药材供试品溶液;或者按照生脉注射液工艺生产,获得制剂供试品溶液。
(2)对照品溶液的制备:
精密称取人参皂苷rg1、re和rb1对照品适量,加20%乙腈配制成浓度分别为0.10mg/ml、0.08mg/ml和0.20mg/ml的溶液,即得。
(3)利用hplc测定的色谱条件:
waterssymmetryshieldtmrp18色谱柱,4.6mm×250mm,5.0μm;检测波长203nm,流动相流速为1.0ml·min-1,柱温为30℃;流动相a为乙腈,流动相b为水,流动相a与b采用梯度洗脱,洗脱梯度如下:
0min时,流动相a:流动相b=0:100,
30min时,流动相a:流动相b=10:90,
40min时,流动相a:流动相b=23:77,
50min时,流动相a:流动相b=23:77,
85min时,流动相a:流动相b=60:40,
95min时,流动相a:流动相b=100:0,
105min时,流动相a:流动相b=100:0,
115min时,流动相a:流动相b=0:100,
在上述色谱条件下,精密吸取供试品溶液10μl进样量,进行高效液相色谱分析,得到红参药材或其制剂的指纹图谱;用同样方法测试对照品溶液。
结果如图1和2所示。
结果分析:
(1)利用hplc方法检测红参药材指纹图谱,以人参皂苷rg1峰面积的5%积分,总共有22个特征峰,其中有9个特征峰指认如下:#5峰为五-羟甲基糠醛、#6峰为核苷、#7峰为人参皂苷rg1、#8峰为人参皂苷re、#9峰为人参皂苷rf、#10峰为人参皂苷rb1、#11峰为人参皂苷rc、#12峰为人参皂苷rb2、#13峰为人参皂苷rd。
(2)以人参皂苷rb1为参照峰,红参药材22个特征峰的相对保留时间分别为:0.092~0.097(#1)、0.122~0.127(#2)、0.168~0.172(#3)、0.185~0.190(#4)、0.195~0.200(#5)、0.298~0.302(#6)、0.825~0.830(#7)、0.840~0.845(#8)、0.993~0.998(#9)、1(#10参照峰)、1.012~1.016(#11)、1.020~1.025(#12)、1.050~1.055(#13)、1.173~1.178(#14)、1.190~1.195(#15)、1.198~1.202(#16)、1.238~1.243(#17)、1.285~1.290(#18)、1.295~1.300(#19)、1.413~1.418(#20)、1.468~1.472(#21)、1.495~1.450(#22);以人参皂苷rb1为参照峰,22个特征峰的相对峰面积分别为:0.300~0.305(#1)、0.613~0.619(#2)、0.118~0.123(#3)、0.068~0.073(#4)、0.490~0.495(#5)、1.220~1.225(#6)、0.870~0.875(#7)、0.575~0.580(#8)、0.458~0.463(#9)、1(#10参照峰)、0.685~0.690(#11)、0.610~0.615(#12)、0.523~0.528(#13)、0.115~0.120(#14)、0.070~0.075(#15)、0.055~0.060(#16)、0.192~0.197(#17)、0.103~0.108(#18)、0.257~0.262(#19)、0.612~0.617(#20)、0.910~0.915(#21)、0.343~0.348(#22)。
(3)红参制剂生脉注射液的指纹图谱中,以人参皂苷rb1为参照峰,17个特征峰的相对保留时间分别为:0.153~0.158(#1)、0.175~0.180(#2)、0.195~0.200(#3)、0.280~0.285(#4)、0.798~0.803(#5)、0.808~0.813(#6)、0.990~0.995(#7)、1(#8参照峰)、1.010~1.015(#9)、1.020~1.025(#10)、1.050~1.055(#11)、1.163~1.168(#12)、1.208~1.213(#13)、1.240~1.245(#14)、1.288~1.293(#15)、1.294~1.299(#16)、1.428~1.433(#17);以人参皂苷rb1为参照峰,17个特征峰的相对峰面积分别为:1.254~1.259(#1)、1.443~1.448(#2)、1.381~1.386(#3)、1.744~1.749(#4)、0.797~0.802(#5)、0.443~0.448(#6)、0.330~0.335(#7)、1(#8参照峰)、0.562~0.567(#9)、1~(#10)、0.446~0.451(#11)、0.288~0.293(#12)、0.068~0.073(#13)、0.383~0.388(#14)、0.437~0.442(#15)、0.196~0.201(#16)、0.506~0.511(#17)。
(4)红参药材中特征峰#5~#13、#19~#22与生脉注射液中特征峰#3~#11、#14~#17相对应。
实施例2
(1)供试品溶液的制备:
制备红参药材(来源与实施例1相同)提取液,精密称定2g红参,加入20ml90%乙醇,回流4小时,过滤,收集续滤液作为药材供试品溶液;或者按照生脉注射液工艺生产(来源与实施例1相同),获得制剂供试品溶液。
(2)对照品溶液的制备:
精密称取人参皂苷rg1、re和rb1对照品适量,加20%乙腈配制成浓度分别为0.10mg/ml、0.08mg/ml和0.20mg/ml的溶液,即得。
(3)利用hplc测定的色谱条件:
waterssymmetryshieldtmrp18色谱柱,4.6mm×250mm,5.0μm;检测波长210nm,流动相流速为1.5ml·min-1,柱温为35℃;流动相a为乙腈,流动相b为水,流动相a与b采用梯度洗脱,洗脱梯度如下:
0min时,流动相a:流动相b=0:100,
30min时,流动相a:流动相b=10:90,
40min时,流动相a:流动相b=23:77,
50min时,流动相a:流动相b=23:77,
85min时,流动相a:流动相b=60:40,
95min时,流动相a:流动相b=100:0,
105min时,流动相a:流动相b=100:0,
115min时,流动相a:流动相b=0:100,
在上述色谱条件下,精密吸取供试品溶液15μl进样量,进行高效液相色谱分析,得到红参药材或其制剂的指纹图谱;用同样方法测试对照品溶液。
实施例3
(1)供试品溶液的制备:
制备红参药材(来源与实施例1相同)提取液,精密称定2g红参,加入20ml90%乙醇,回流4小时,过滤,收集续滤液作为药材供试品溶液;或者按照生脉注射液工艺生产(来源与实施例1相同),获得制剂供试品溶液。
(2)对照品溶液的制备:
精密称取人参皂苷rg1、re和rb1对照品适量,加20%乙腈配制成浓度分别为0.10mg/ml、0.08mg/ml和0.20mg/ml的溶液,即得。
(3)利用hplc测定的色谱条件:
waterssymmetryshieldtmrp18色谱柱,4.6mm×250mm,5.0μm;检测波长200nm,流动相流速为0.5ml·min-1,柱温为25℃;流动相a为乙腈,流动相b为水,流动相a与b采用梯度洗脱,洗脱梯度如下:
0min时,流动相a:流动相b=0:100,
30min时,流动相a:流动相b=12:88,
40min时,流动相a:流动相b=23:77,
50min时,流动相a:流动相b=23:77,
85min时,流动相a:流动相b=60:40,
95min时,流动相a:流动相b=100:0,
105min时,流动相a:流动相b=100:0,
115min时,流动相a:流动相b=0:100,
在上述色谱条件下,精密吸取供试品溶液8μl进样量,进行高效液相色谱分析,得到红参药材或其制剂的指纹图谱;用同样方法测试对照品溶液。
分别以实施例1至3建立的指纹图谱为标准,评价同样参药材样品的质量,显示三种评价方法的结果一致,均达到95%以上的相似度,说明三种方法都可以用于评价红参药材的质量。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 还没有人留言评论。精彩留言会获得点赞!