一种原油中破乳剂浓度的检测方法与流程
本发明属于原油领域,涉及一种原油中破乳剂浓度的检测方法。
背景技术:
目前油田现场亟需测定原油中破乳剂的浓度,以指导现场实际加药量,同时控制成本。但迄今为止,还未有一种快速、经济的测定原油中破乳剂含量的方法。由于油田现场使用的破乳剂主要为聚氧乙烯基非离子表面活性剂,因此,本专利仅对此提供一种快速、经济的间接检测原油中破乳剂含量的测定方法。
该专利方法原理是利用配位反应产生的颜色变化,采用分光光度计测定测量配体含量,依此反应,测定油田污水中破乳剂的浓度,同时根据现场破乳剂总加量,进而计算得知原油中破乳剂的含量。
当聚氧乙烯型表面活性剂存在时,聚氧乙烯链就会取代硫氰酸钴co(scn)3·3h20中的水分子,其醚氧原子与co2+直接配位,co2+的配位数变为4,呈现深蓝色,萃取后可用于分光光度法测定。
对目前国内外表面活性剂的测定方法进行了系统详细的调研:
目前非离子表面活性剂的测定方法主要为离子选择电极法(ise)、气相色谱-质谱法(gc-ms)、液相色谱-质谱法(hplc-ms)、毛细管电泳法(ce)、超临界流体色谱法(sfc)、紫外分光光度法(uv)等。
1)色谱法
色谱法能有效分离并检测混合表面活性剂,该技术分析速度快、成本低、分离度及灵敏度高。液相色谱法测定非离子表面活性剂的报道相对较多。
2013年,zhang等建立了一种快速高效液相色谱法测定吐温80及其分解物,9次测定的平均回收率为95.12%,说明该方法准确度较高,但ri需要洗脱,削弱了分离能力。
2)质谱法
质谱分析是通过测定离子质荷比的一种分析方法。不含eo链的非离子表面活性剂在质谱仪上没有信号。2010年,beneito-cambra等为了解决这些问题,在这类物质末端-oh接上铬(vi)的氧化物,再用乙酸乙酯萃取。实验发现ae的检测信号被加强可进行测定。质谱仪不能直接测定的非离子表面活性剂,可采用间接法测定。
3)色谱-质谱联用
①液相色谱-质谱法
质谱仪自身具有独特优势,其接口能与多种仪器相匹配,另外质谱仪不适宜测定混合表面活性剂,因此液相色谱-质谱联用为非离子表面活性剂测定提供了更高效的方法。2006年,pablo等首次采用液相色谱-质谱法(lc-ms)同时测定环境水和沉积物中的阴离子表面活性剂(烷基苯磺酸盐、烷基硫酸盐、乙氧基化烷基硫酸钠)和非离子表面活性剂(壬基酚聚氧乙烯醚与聚醚多元醇)及它们的代谢物。后来发展的液相色谱-串联质谱法(lc-ms/ms)可以同时进行定性和定量分析。
②气相色谱-质谱法
气相色谱-质谱法(gc-ms)的分离能力在某些情况下比hplc-ms强。gc-ms适宜测定挥发性较强的非离子表面活性剂(eo链较短)。硅烷化非离子表面活性剂宜用气相色谱法测定。
对于复杂混合体系,2010年,wulf等采用全二维气相色谱-质谱法测定工业用清洗剂中的非离子、阴离子与部分阳离子表面活性剂。该方法在传统gc基础上提高了混合物的分辨率,增大了表面活性剂的峰容量。
4)电化学法
选择性电极电位法已用来研究测定表面活性剂。目前主要基于在金属离子存在下,非离子表面活性剂能与四苯硼钠反应得到活性物,再通过制备离子选择性电极测定目标物。1965年,levins等首次将这种沉淀反应应用到非离子表面活性剂电位滴定上。对于膜电极,kulapina等在2000年从电化学、动力学和传递性质等几个方面进行了研究,提供了该类电极的基础理论知识。
2007年,sak-bosnar等采用ba2+-ns-tpb混合物作为活性物制备聚氯乙烯膜非离子表面活性剂选择性电极。其中非离子表面活性剂选取eo数为80的聚醚。将制备电极用于测定含不同个数eo的非离子表面活性剂,四苯硼钠为滴定剂。2009年,sak-bosnar等对pvc膜电极进行创新。
2001年,martinez-barrachina等将该方法与离子选择电极技术联用,方便且适合连续测定目标物,提高了测定非离子表面活性剂的效率。
2007年,bas等将该技术应用于测定非离子表面活性剂曲拉通x-100,回收率在94~103%之间,表明该方法准确度较高。
5)光谱法
光谱法具有灵敏度高、选择性好、准确度高、分析成本低、操作简便且快速等优点。
①红外光谱法
对于复杂的混合物,衰减全反射傅里叶变换红外光谱法(atr-ftir)是一种有用的分析工具。2005年,carolei等采用该技术直接、快速地同时测定洗手液和洗发水中的阴离子、两性与非离子表面活性剂。该测定不需要稀释,对样品也不需要预处理。2008年,kargosha等采用atr-ftir联合多元线性回归法同时测定未经稀释的洗手液中的一种非离子表面活性剂椰子油脂肪酸二乙醇酰胺、两种阴离子表面活性剂十二烷基磺酸钠和直链烷基磺酸盐,并记录波长在1305~990cm-1之间的数据。
②化学发光法
化学发光法具有操作简单、检出限低、检测区间宽、被测时间短等优点,将它应用到非离子表面活性剂的测定是一个重要创新。2009年,liu等首次采用化学发光法测定湖水中的非离子表面活性剂曲拉通x-100。
③分光光度法
由于多数非离子表面活性剂自身不具有紫外吸收,因此常采用显色试剂测定该类物质。钴盐比色法定量分析非离子表面活性剂的研究较早,随后favretto等利用苦味酸盐,tei等采用四溴酚酞乙酯钾盐,crisp等利用硫氰锌化钾,文献利用碘化物与非离子表面活性剂反应测定该物质含量。
除了以上介绍的几种主要测定非离子表面活性剂方法,还有毛细管电泳分析法、荧光光谱法、免疫酶测定法等。
在这些方法中,重量法操作复杂、准确度不高,比色法精确度不高,色谱法受仪器限制。
色谱法:该方法需要等待洗脱过程,削弱了分离能力。elsd测定aeo的检测范围较窄,且色谱图得到很多峰,需要通过标准物质逐一比对。此外,该方法最大的缺点是不能快速、方便地进行现场测定。
质谱法:(α-cd)能与非离子表面活性剂曲拉通x-100产生络合作用,质谱图中只能得到[α-cd-h]峰,不能得到α-cd与曲拉通x-100反应的络合物。
电化学法:该方法准确度较高,但其检测范围较窄,质量浓度在0.05~20mg/l之间,且电位值会漂移,因此该测定方法不稳定且重复性差。
光谱法:光谱法具有灵敏度高、但对于多组分化学性质较相似的混合物,因为它们的光谱峰会相互重叠严重干扰定量分析,因此采用光谱分析难度较大。
因此,开发一种灵敏度高、准确性好、测试方法简单、成本低且检测范围广的检测方法很有必要。
技术实现要素:
本发明的目的在于提供一种原油中破乳剂浓度的检测方法,所述检测方法灵敏度高、准确性好、测试工艺简单、成本低且适用范围广。
为达此目的,本发明采用以下技术方案:
本发明的目的在于提供一种原油中破乳剂浓度的检测方法,所述检测方法包括如下步骤:
(1)利用破乳剂配制浓度梯度的标准溶液,加入萃取剂萃取,显色剂显色,通过紫外分光光度法测定标准溶液有机层的吸光度,根据吸光度和浓度绘制标准曲线;
(2)在待测样中加入已知含量的破乳剂,将待测样中油相和水相分离,得到水相层和原油层;
(3)将步骤(2)得到的水相层中加入萃取剂萃取和显色剂显色,通过紫外分光光度法测定待测样有机层的吸光度,根据步骤(1)绘制的标准曲线定量步骤(2)得到的水相中破乳剂的浓度;
(4)根据步骤(3)得到的水相层中破乳剂的浓度和步骤(2)中的已知含量的破乳剂,得到油相中的破乳剂浓度。
本发明采用紫外分光光度法对破乳剂进行检测,检测方法简单,原料易得,不需要使用比较昂贵的仪器;检测灵敏度较高,准确性好,相对标准偏差较低,重复性好,适用范围广。
本发明在待测样中加入破乳剂之前,先对待测样水相中的破乳剂的含量进行检测。
本发明根据需要选用不同浓度梯度的标准溶液,相邻浓度间相差不大,且涵盖范围较广,绘制的标准曲线的线性较好,准确率较高。
本发明配置不同浓度的标准溶液时,采用移液管进行移取,可保证较小的误差。
在本发明中,所述萃取剂为二氯甲烷和/或三氯甲烷,优选二氯甲烷。
本发明选用萃取剂为二氯甲烷或三氯甲烷,虽然二者均有较好的萃取效果,但是三氯甲烷的毒性较大,因此优选二氯甲烷。
在本发明中,所述萃取剂的浓度不低于99.5%,例如99.5%、99.55%、99.6%、99.65%、99.7%、99.75%、99.8%、99.85%、99.9%、99.95%、100%等。
在本发明中,所述萃取剂的萃取时间为10-12min,例如10min、10.2min、10.4min、10.6min、10.8min、11min、11.2min、11.4min、11.6min、11.8min、12min等,优选10min。
本发明选取的萃取时间为10-12min,随着萃取剂加入时间的增加,吸光度逐渐增加,当萃取时间为10min时,随萃取时间增加,吸光度趋于稳定;因为络合物中有机成分与萃取剂的结合速率较高,最优选的萃取时间为10min。
在本发明中,所述萃取剂的添加体积为5-25ml,例如5ml、7ml、10ml、12ml、15ml、18ml、20ml、22ml、25ml等,优选10ml。
本发明中,随着萃取剂含量的增加,吸光度会降低,因为在一定温度情况下,萃取分配系数等于萃取平衡时有机相与水相中物质浓度之比,该比值为定值。因此改变萃取剂的体积,会改变相应平衡时萃取物的浓度。
在本发明中,所述显色剂为硫氰酸钴盐溶液。
本发明选用的显色剂为硫氰酸钴盐溶液,硫氰酸钴盐溶液中的co(scn)42-n的离子可与破乳剂中聚氧乙烯基发生配位反应,生成络合物,会发生颜色的变化,通过测定钴离子的含量,即可测得配体的含量,从而测得破乳剂的含量。
在本发明中,所述硫氰酸钴盐溶液的配置方法为:称取3g硝酸钴、20g硫氰酸铵和20g氯化钾溶于蒸馏水中,搅拌均匀,转移至100ml容量瓶中,用蒸馏水定容,摇匀,得到硫氰酸钴盐溶液。
在本发明中,所述硝酸钴、硫氰酸铵和氯化钾均为分析纯。
在本发明中,所述硫氰酸钴盐溶液的添加体积为5-9ml,例如5ml、5.5ml、6ml、6.5ml、7ml、7.5ml、8ml、8.5ml、9ml等,优选5ml。
在本发明中,所述显色剂的显色时间为10-18min,例如10min、11min、12min、13min、14min、15min、16min、17min、18min等,优选10min。
在本发明中,所述吸光度为波长在600-700nm(例如600nm、610nm、620nm、630nm、640nm、650nm、660nm、670nm、680nm、690nm、700nm等)的吸光度。
优选地,所述波长为640nm。
在本发明中,二氯甲烷和三氯甲烷均在600-700nm处出现吸收峰,在用相对应的紫外分光光度计时,选择的波长为640nm,在640nm时,紫光效应最强。
在本发明中,所述显色剂显色温度为15-30℃,例如15℃、18℃、20℃、22℃、25℃、27℃、30℃等。
在本发明中,所述紫外分光光度法的测试温度为15-30℃,例如15℃、18℃、20℃、22℃、25℃、27℃、30℃等。
本发明控制显色剂的显色温度和紫外分光光度法的测试温度为15-30℃,是因为萃取剂若选用二氯甲烷,二氯甲烷的沸点较低,若采用较高温度,会造成二氯甲烷的挥发,造成测试结果不准确。
在本发明中,步骤(2)所述待测样为油田试样。
在本发明中,步骤(3)所述水相层的体积为100ml。
实验用水样为油田试样。若取回水样含油较多,则需要进行油水分离,将油相与水相充分分离,但不可引入其他破乳剂,可将样品升温,并加入适量nacl(4.00g/l)进行破乳与分层。
在本发明中,所述待测样的体积为100ml。
在本发明中,所述破乳剂包括聚乙二醇、聚氧乙烯醚或聚氧乙烯-聚氧丙烯共聚醚中的任意一种或至少两种的组合。
在本发明中,所述紫外分光光度法是通过紫外分光光度计进行测量的。
在本发明中,所述紫外分光光度计包括uv-1800。
在本发明中,所述uv-1800的比色皿尺寸为1cm×1cm×5cm。
作为本发明的优选方案,所述检测方法包括如下步骤:
(1)称取3g硝酸钴、20g硫氰酸铵和20g氯化钾溶于蒸馏水中,搅拌均匀,转移至100ml容量瓶中,用蒸馏水定容,摇匀,得到硫氰酸钴盐溶液;利用破乳剂配制浓度梯度的标准溶液,加入体积为10-25ml浓度为99.5%的二氯甲烷或三氯甲烷萃取10-12min,而后加入体积为5-9ml的步骤(1)得到的硫氰酸钴盐溶液在15-30℃显色10-18min,利于紫外分光光度计在测试温度为15-30℃时通过紫外分光光度法测定标准溶液有机层在波长为600-700nm的吸光度,根据吸光度和浓度绘制标准曲线;
(2)在待测样中加入已知含量的破乳剂,将待测样中油相和水相分离,得到水相层和原油层;
(3)将步骤(2)得到的100ml的水相层中加入体积为10-25ml浓度为99.5%的二氯甲烷或三氯甲烷萃取10-12min,而后加入体积为5-9ml的步骤(1)得到的硫氰酸钴盐溶液在15-30℃显色10-18min,利于紫外分光光度计在测试温度为15-30℃时通过紫外分光光度法测定待测样有机层在波长为600-700nm的吸光度,根据步骤(1)绘制的标准曲线定量步骤(2)得到的水相中破乳剂的浓度;
(4)根据步骤(3)得到的水相中破乳剂的浓度和步骤(2)中的已知含量的破乳剂,得到油相中的破乳剂浓度。
相对于现有技术,本发明具有以下有益效果:
本发明采用紫外分光光度法对原油中破乳剂浓度进行测定,该检测方法检测工艺简单,效率较高,且不需要特别昂贵的仪器就可以达到;该方法具有较高的精确度,相对标准偏差较小,准确率较高;本发明选用破乳剂作标准曲线,避免使用单一的标准物质制备标准曲线会造成结果不准确的问题。
附图说明
图1是本发明实施例1中雁一联合站破乳剂吸光度和浓度的标准曲线;
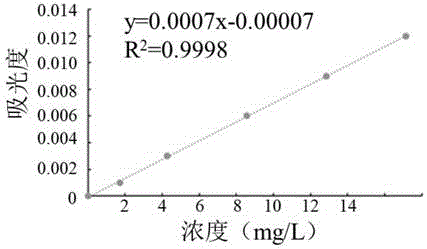
图2是本发明实施例2中任一联合站破乳剂吸光度和浓度的标准曲线;
图3是本发明实施例3中马一联合站破乳剂吸光度和浓度的标准曲线。
具体实施方式
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
实施例1
本实施例选用雁一联合站为测试目标,在于提供一种原油中聚氧乙烯基浓度的检测方法,所述检测方法包括如下步骤:
(1)称取3g硝酸钴、20g硫氰酸铵和20g氯化钾溶于蒸馏水中,搅拌均匀,转移至100ml容量瓶中,用蒸馏水定容,摇匀,得到硫氰酸钴盐溶液;利用破乳剂配制浓度梯度分别为0mg/l、5mg/l、10mg/l、15mg/l、20mg/l的标准溶液,加入体积为10ml浓度为99.5%的二氯甲烷或三氯甲烷萃取10min,而后加入体积为5ml的步骤(1)得到的硫氰酸钴盐溶液在25℃显色10min,利于紫外分光光度计在测试温度为25℃时通过紫外分光光度法测定标准溶液有机层在波长为640nm的吸光度,根据吸光度和浓度绘制标准曲线;
(2)在9000立方米待测样(质量含水率91.11%)中加入125kg的破乳剂(此时破乳剂浓度为13.89mg/l),将待测样中油相和水相分离,得到水相层和原油层;
(3)将步骤(2)得到的100ml的水相层中加入体积为10ml浓度为99.5%的二氯甲烷或三氯甲烷萃取10min,而后加入体积为5ml的步骤(1)得到的硫氰酸钴盐溶液在25℃显色10min,利于紫外分光光度计在测试温度为25℃时通过紫外分光光度法测定待测样有机层在波长为640nm的吸光度,根据步骤(1)绘制的标准曲线定量步骤(2)得到的水相中破乳剂的浓度;
(4)根据步骤(3)得到的水相中破乳剂的浓度及步骤(2)中的待测样中的破乳剂含量、含水率即可计算出油相中的破乳剂浓度。
图1为本实施例吸光度和破乳剂浓度的标准曲线图;根据吸光度和浓度作标准曲线,得到回归方程y=0.0007x+0.0002,由回归方程得到线性相关系数(r2)为0.9912,说明该标准曲线线性良好。
以该标准曲线为依据,对雁一联合站原油中破乳剂浓度进行测试,测试结果为水相中破乳剂的浓度为5.4mg/l;则原油中破乳剂的浓度为17.6mg/l;
其中:水密度为1g/cm3,原油密度为0.81g/cm3,待测样中质量含水率为91.11%;
其中:加入破乳剂的质量为125kg,水相中破乳剂的浓度为5.4mg/l,待测样体积为9000立方米,待测样体积含水率为上述计算的待测样的体积含水率。
将该原油中的破乳剂用气质联用分析测定,测得原油中破乳剂的浓度为17.9mg/l;本实施例的检测结果与气质联用测得的结果的相对误差为1.68%,证明本发明的测试方法准确可靠。
实施例2
本实施例选用任一联合站为测试目标,在于提供一种原油中聚氧乙烯基浓度的检测方法,所述检测方法包括如下步骤:
(1)称取3g硝酸钴、20g硫氰酸铵和20g氯化钾溶于蒸馏水中,搅拌均匀,转移至100ml容量瓶中,用蒸馏水定容,摇匀,得到硫氰酸钴盐溶液;利用破乳剂配制浓度梯度分别为2mg/l、4mg/l、9mg/l、13mg/l、18mg/l的标准溶液,加入体积为10ml浓度为99.5%的二氯甲烷或三氯甲烷萃取10min,而后加入体积为5ml的步骤(1)得到的硫氰酸钴盐溶液在25℃显色10min,利于紫外分光光度计在测试温度为25℃时通过紫外分光光度法测定标准溶液有机层在波长为640nm的吸光度,根据吸光度和浓度绘制标准曲线;
(2)在18000立方米待测样(质量含水率94.44%)中加入96kg的破乳剂(此时破乳剂浓度为5.33mg/l),将待测样中油相和水相分离,得到水相层和原油层;
(3)将步骤(2)得到的100ml的水相层中加入体积为10ml浓度为99.5%的二氯甲烷或三氯甲烷萃取10min,而后加入体积为1-9ml的步骤(1)得到的硫氰酸钴盐溶液在25℃显色10min,利于紫外分光光度计在测试温度为25℃时通过紫外分光光度法测定待测样有机层在波长为640nm的吸光度,根据步骤(1)绘制的标准曲线定量步骤(2)得到的水相中破乳剂的浓度;
(4)根据步骤(3)得到的水相中破乳剂的浓度及步骤(2)中的待测样中的破乳剂含量、含水率即可计算出油相中的破乳剂浓度。
图2为本实施例吸光度和破乳剂浓度的标准曲线图;根据吸光度和浓度作标准曲线,得到回归方程y=0.0007x-0.00007,由回归方程得到线性相关系数(r2)为0.9998,说明该标准曲线线性良好。
以该标准曲线为依据,对任一联合站原油中破乳剂浓度进行测试,测试结果为水相中破乳剂的浓度为3.0mg/l;则原油中破乳剂的浓度为27.6mg/l。
将该原油中的破乳剂用气质联用分析测定,测得原油中破乳剂的浓度为27.2mg/l;本实施例的检测结果与气质联用测得的结果的相对误差为1.47%,证明本发明的测试方法准确可靠。
实施例3
本实施例选用马一联合站为测试目标,在于提供一种原油中聚氧乙烯基浓度的检测方法,所述检测方法包括如下步骤:
(1)称取3g硝酸钴、20g硫氰酸铵和20g氯化钾溶于蒸馏水中,搅拌均匀,转移至100ml容量瓶中,用蒸馏水定容,摇匀,得到硫氰酸钴盐溶液;利用破乳剂配制浓度梯度分别为12mg/l、25mg/l、35mg/l、50mg/l、100mg/l、170mg/l、220mg/l的标准溶液,加入体积为10ml浓度为99.5%的二氯甲烷或三氯甲烷萃取10min,而后加入体积为5ml的步骤(1)得到的硫氰酸钴盐溶液在25℃显色10min,利于紫外分光光度计在测试温度为25℃时通过紫外分光光度法测定标准溶液有机层在波长为640nm的吸光度,根据吸光度和浓度绘制标准曲线;
(2)在1100立方米待测样(质量含水率91.67%)中加入10kg的破乳剂(此时破乳剂浓度为9.09mg/l),将待测样中油相和水相分离,得到水相层和原油层;
(3)将步骤(2)得到的100ml的水相层中加入体积为10ml浓度为99.5%的二氯甲烷或三氯甲烷萃取10min,而后加入体积为1-9ml的步骤(1)得到的硫氰酸钴盐溶液在25℃显色10min,利于紫外分光光度计在测试温度为25℃时通过紫外分光光度法测定待测样有机层在波长为640nm的吸光度,根据步骤(1)绘制的标准曲线定量步骤(2)得到的水相中破乳剂的浓度;
(4)根据步骤(3)得到的水相中破乳剂的浓度及步骤(2)中的待测样中的破乳剂含量、含水率即可计算出油相中的破乳剂浓度。
图3为本实施例吸光度和破乳剂浓度的标准曲线图;根据吸光度和浓度作标准曲线,得到回归方程y=0.0007x+0.0002,由回归方程得到线性相关系数(r2)为0.9912,说明该标准曲线线性良好。
以该标准曲线为依据,对马一联合站原油中破乳剂浓度进行测试,测试结果为水相中破乳剂的浓度为2.4mg/l;则原油中破乳剂的浓度为21.3mg/l。
将该原油中的破乳剂用气质联用分析测定,测得原油中破乳剂的浓度为21.0mg/l;本实施例的检测结果与气质联用测得的结果的相对误差为1.43%,证明本发明的测试方法准确可靠。
实施例4
本实施例选用雁一联合站为测试目标,在于提供一种原油中聚氧乙烯基浓度的检测方法,所述检测方法包括如下步骤:
(1)称取3g硝酸钴、20g硫氰酸铵和20g氯化钾溶于蒸馏水中,搅拌均匀,转移至100ml容量瓶中,用蒸馏水定容,摇匀,得到硫氰酸钴盐溶液;
(2)利用破乳剂配制浓度梯度分别为0mg/l、5mg/l、10mg/l、15mg/l、20mg/l的标准溶液,加入体积为25ml浓度为99.5%的二氯甲烷或三氯甲烷萃取12min,而后加入体积为9ml的步骤(1)得到的硫氰酸钴盐溶液在30℃显色18min,利于紫外分光光度计在测试温度为30℃时通过紫外分光光度法测定标准溶液有机层在波长为640nm的吸光度,根据吸光度和浓度绘制标准曲线;
(2)在9000立方米待测样(质量含水率91.11%)中加入125kg(此时破乳剂浓度为13.89mg/l)的破乳剂,将待测样中油相和水相分离,得到水相层和原油层;
(3)将步骤(2)得到的100ml的水相层中加入体积为25ml浓度为99.5%的二氯甲烷或三氯甲烷萃取12min,而后加入体积为9ml的步骤(1)得到的硫氰酸钴盐溶液在30℃显色18min,利于紫外分光光度计在测试温度为30℃时通过紫外分光光度法测定待测样有机层在波长为640nm的吸光度,根据步骤(1)绘制的标准曲线定量步骤(2)得到的水相中破乳剂的浓度;
(4)根据步骤(3)得到的水相中破乳剂的浓度及步骤(2)中的待测样中的破乳剂含量、含水率即可计算出油相中的破乳剂浓度。
以实施例1得到的标准曲线为依据,对雁一联合站原油中破乳剂浓度进行测试,测试结果为水相中破乳剂的浓度为2.9mg/l;则原油中破乳剂的浓度为17.4mg/l。
将该原油中的破乳剂用气质联用分析测定,测得原油中破乳剂的浓度为17.9mg/l;本实施例的检测结果与气质联用测得的结果的相对误差为2.76%,证明本发明的测试方法准确可靠。
实施例5
与实施例1的区别仅在于硫氰酸钴盐溶液的添加体积为9ml,其余原料与检测方法均与实施例1相同。
本实施例的检测结果与气质联用测得的结果的相对误差为2.76%,证明本发明的测试方法准确可靠。
实施例6
与实施例1的区别仅在于显色时间为18min,其余原料与检测方法均与实施例1相同。
本实施例的检测结果与气质联用测得的结果的相对误差为1.78%,证明本发明的测试方法准确可靠。
实施例7
与实施例1的区别仅在于萃取剂为三氯甲烷,其余原料与检测方法均与实施例1相同。
本实施例的检测结果与气质联用测得的结果的相对误差为2.02%,证明本发明的测试方法准确可靠,但是三氯甲烷毒性较大,不利于工业应用。
实施例8
与实施例1的区别仅在于二氯甲烷的体积为25ml,其余原料与检测方法均与实施例1相同。
本实施例的检测结果与气质联用测得的结果的相对误差为1.12%,证明本发明的测试方法准确可靠。
实施例9
与实施例1的区别仅在于萃取时间为12min,其余原料与检测方法均与实施例1相同。
本实施例的检测结果与气质联用测得的结果的相对误差为1.68%,证明本发明的测试方法准确可靠。
对比例1
与实施例1的区别仅在于硫氰酸钴盐溶液的添加体积为1ml,其余原料与检测方法均与实施例1相同。
本对比例的检测结果与气质联用测得的结果的相对误差为6.20%,证明本发明的测试方法准确率较低。
对比例2
与实施例1的区别仅在于显色时间为2min,其余原料与检测方法均与实施例1相同。
本对比例的检测结果与气质联用测得的结果的相对误差为10.12%,证明本发明的测试方法准确率较低。
对比例3
与实施例1的区别仅在于二氯甲烷的体积为5ml,其余原料与检测方法均与实施例1相同。
本对比例的检测结果与气质联用测得的结果的相对误差为5.46%,证明本发明的测试方法准确率较低。
对比例4
与实施例1的区别仅在于显色温度为40℃,其余原料与检测方法均与实施例1相同。
本对比例的检测结果与气质联用测得的结果的相对误差为13.12%,证明本发明的测试方法准确率较低。
对比例5
与实施例1的区别仅在于萃取时间为2min,其余原料与检测方法均与实施例1相同。
本对比例的检测结果与气质联用测得的结果的相对误差为8.84%,证明本发明的测试方法准确率较低。
申请人声明,以上所述仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,所属技术领域的技术人员应该明了,任何属于本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!