一种核级金属钠中微量氯离子含量的检测方法与流程
本发明属于示范快堆化学分析技术领域,具体涉及一种核级金属钠中微量氯离子含量的检测方法。
背景技术:
电解氯化钠制造金属钠的工艺过程中会不可避免地引进微量氯。钠中氯常以氯化物形式存在,它对钠冷快中子反应堆的包壳材料和结构材料有腐蚀作用,因此作为钠冷快中子反应堆冷却剂的核级钠,氯的含量需控制在30μg/g以下。为此,必须建立一种钠中氯的测定方法,以对供货和入堆钠进行质量监控。然而氯的化学性质活泼,几乎存在于所有的试剂和材料中,实验中容易引入或丢失,所以微量氯的分析有一定的难度。金属钠中微量氯的分析难度更大,也较难找到满意的分析方法。文献资料《核级金属钠中微量氯的分析》公开了一种采用真空蒸馏-分光光度法测定钠中氯含量的方法,该分析方法耗时长、操作繁琐,且具有一定危险性。因此,迫切需要一种简便易操作而又高效的核级金属钠中微量氯离子含量的检测方法,能够快速、准确又安全的分析核级金属钠中微量氯离子含量。
技术实现要素:
本发明的目的在于针对现有金属钠中氯化物检测方法耗时长、操作繁琐的问题,提供了一种利用离子色谱的核级金属钠中微量氯离子含量的检测方法。
本发明的技术方案如下:
本发明提供了一种核级金属钠中微量氯离子含量的检测方法,依次包括如下步骤:
s1.称取金属钠样品,加入无水乙醇中将样品溶解,然后加入高纯水定容,得到样品溶液s,然后采用离子色谱法检测得到样品溶液s中的氯离子含量;
s2.配制空白溶液,即在处理时不加金属钠样品,其余与步骤s1一致,得到空白溶液的氯离子浓度;
s3.使用带有基体中和模块和基体消除装置的离子色谱仪检测s1配制的样品溶液s中的氯离子含量;其中,离子色谱的淋洗液e为碳酸钠和碳酸氢钠的混合溶液,碳酸钠浓度为2.5-3.8mmol/l,碳酸氢钠浓度为0.8-1.2mmol/l;抑制器再生液r1为磷酸水溶液,浓度范围50-200mmol/l;基体中和模块再生液r2为草酸溶液,浓度范围50-200mmol/l;
s4.使用带有基体中和模块和基体消除装置的离子色谱仪检测s2配制的空白溶液中的氯离子含量;
s5.通过加标回收率对仪器测量的准确度进行检验,配制氯离子标准溶液加入样品溶液s中,使用带有基体中和模块和基体消除装置的离子色谱仪检测溶液中的氯离子含量;
s6.计算金属钠样品中的氯含量,用步骤1得到的样品溶液s的氯离子浓度和步骤2的得到的空白溶液的氯离子浓度之差,乘以样品溶液s的体积,再除以步骤1称取的金属钠样品质量。
进一步地,所述基体中和模块由三个并联的中和仓组成,三个中和仓轮流工作,每处理一个样品轮换一个中和仓。
进一步地,所述基体中和模块再生液r2浓度范围80-100mmol/l。
进一步地,所述离子色谱仪含有淋洗液流路、再生液流路、进样流路和分析检测流路,所述分析检测流路包含依次串联的阴离子交换柱、抑制器和电导检测池。
进一步地,所述步骤3依次包括如下步骤:
s3.1样品溶液s通过进样流路进入离子色谱仪的基体中和模块,样品溶液s中的钠离子和基体中和模块中的氢离子进行交换,乙醇钠和氢氧化钠分别变成乙醇和水;
s3.2基体中和模块流出的溶液经过基体消除装置,把溶液中的乙醇进行去除;
s3.3溶液进入阴离子色谱交换柱,溶液中的氯离子在阴离子交换柱内不断被吸附洗脱,使得氯离子与其他杂质分离,进入抑制器;
s3.4在抑制器中,将淋洗液中的钠离子转换为氢离子,使氯离子的信号值增强,背景电导降低,随后进入电导检测器中检测生成谱图。
进一步地,所述淋洗液e中碳酸钠浓度为3.2mmol/l,碳酸氢钠浓度为1.0mmol/l。
进一步地,所述再生液r1溶液浓度为100mmol/l。
进一步地,所述再生液r2溶液浓度为100mmol/l。
进一步地,所述基体消除装置包括预浓缩柱,载体是超纯水。
经过基体中和模块后,样品溶液s中主要成份有乙醇、氢离子、氯离子和其他阴离子,样品溶液s进入基体消除装置(基体消除装置主要为预浓缩柱),仪器中的超纯水作为载体带着样品溶液s流经预浓缩柱(预浓缩柱为阴离子交换柱),氯离子和其他阴离子被吸附至预浓缩柱上,乙醇、氢离子被超纯水冲走,排废,达到除去乙醇基体的目的。然后将预浓缩柱切换至离子色谱分析检测流路,用淋洗液e将预浓缩柱上氯离子和其他阴离子洗脱下来,实现进样。
本发明的有益效果在于:
本发明所述检测核级金属钠中氯离子含量的方法,首先将样品加无水乙醇和水进行溶解,配制成溶液,使得样品能以溶液形式进入离子色谱仪,使得利用离子色谱方法测金属钠中氯离子含量得以实现。将样品溶液进行基体中和及消除后通过阴离子交换柱,利用各种阴离子与阴离子交换柱的不同亲和力而形成差速迁移,实现氯离子与其他阴离子的分离。采用此方法,通过简便一次操作就能准确获得金属钠中氯离子的检测结果,并通过使用基体中和模块和基体消除装置,降低了检出限,延长了色谱柱的使用时间,操作简便,数据重现性好。相比与之前金属钠中氯离子含量的测量方法,大大降低了前处理的时间和危险性,操作也更加简便。
附图说明
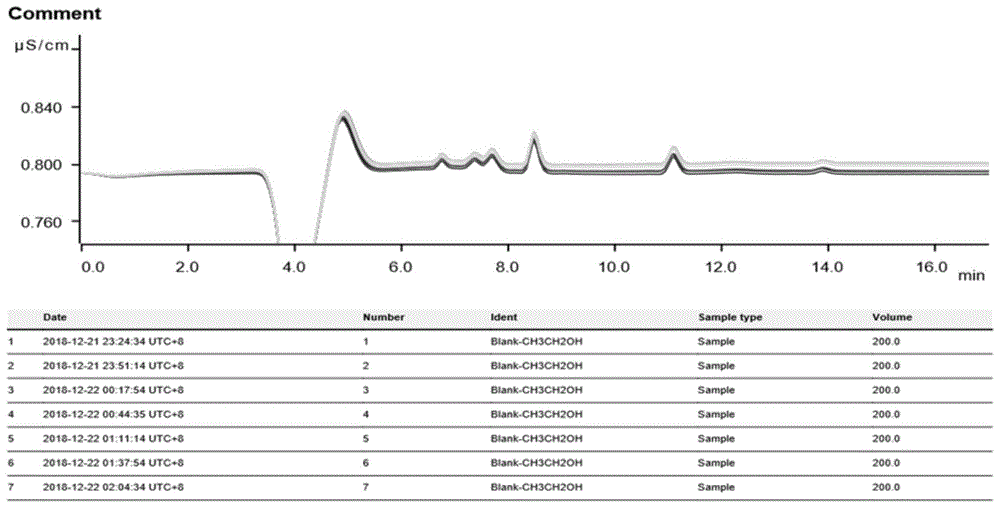
图1是采用实施例2所述方法对实施例中空白溶液稀释10倍后进行连续7次分析的谱图叠加;
图2是采用实施例2所述方法对实施例空白溶液稀释10倍后进行连续7次分析的氯离子含量散点图;
图3是采用实施例2所述方法对实施例空白溶液稀释10倍后进行分析的谱图;
图4是采用本发明所述方法对实施例5#1样品溶液稀释10倍后进行分析的谱图;
图5是采用实施例2所述方法对实施例5#2样品溶液稀释10倍后进行分析的谱图;
图6是采用实施例2所述方法对实施例8#1样品溶液稀释10倍后进行分析的谱图;
图7是采用实施例2所述方法对实施例8#2样品溶液稀释10倍后进行分析的谱图;
图8是采用实施例2所述方法对实施例中氯离子标准溶液(1ppb)进行分析的谱图;
图9是采用实施例2所述方法对实施例中氯离子标准溶液进行分析后绘制的工作曲线;
图10是采用实施例2所述方法对实施例5#1样品溶液稀释10倍后分别加入0ppb、4ppb、8ppb和16ppb氯离子标准溶液进行分析的谱图叠加;
图11是采用实施例2所述方法对实施例8#2样品溶液稀释10倍后分别加入0ppb、4ppb、8ppb和16ppb氯离子标准溶液进行分析的谱图叠加。
具体实施方式
下面结合附图及具体实施例对本发明作进一步详细说明。
实施例1
本实施例提供了一种核级金属钠中微量氯离子含量的检测方法,依次包括如下步骤:
s1.称取金属钠样品,加入无水乙醇中将样品溶解,然后加入高纯水定容,得到样品溶液s,然后将样品溶液s稀释10倍后,采用离子色谱法检测其中的氯离子含量;
s2.配制空白溶液,在处理样品的同时,做空白试验,即在处理时不加金属钠,其余与步骤s1一致;
s3.配制已知浓度的氯离子标准溶液;
s4.使用带有基体中和模块的离子色谱仪检测s1配制的样品溶液s中的氯离子含量;其中,离子色谱的淋洗液e为碳酸钠和碳酸氢钠的混合溶液,碳酸钠浓度为2.5-3.8mmol/l,碳酸氢钠浓度为0.8-1.2mmol/l;抑制器再生液r1为磷酸水溶液,浓度范围50-200mmol/l;基体中和模块再生液r2为草酸水溶液,浓度范围50-200mmol/l;
s4.1样品溶液s通过进样流路进入离子色谱仪的基体中和模块,样品溶液s中的钠离子和基体中和模块中的氢离子进行交换,乙醇钠和氢氧化钠分别变成乙醇和水;
s4.2基体中和模块流出的溶液经过基体消除装置,把溶液中的乙醇进行去除;
s4.3溶液进入阴离子色谱交换柱,溶液中的氯离子在阴离子交换柱内不断被吸附洗脱,使得氯离子与其他杂质分离,进入抑制器;
s4.4在抑制器中,将淋洗液中的钠离子转换为氢离子,使氯离子的信号值增强,背景电导降低,随后进入电导检测器中检测生成谱图。
基体中和模块由三个并联的中和仓组成,三个中和仓轮流工作,每处理一个样品轮换一个中和仓。基体中和过程,三个中和仓中的一个在工作,第二个再生,第三个冲洗,保证中和仓容量与样品处理的连续性。在处理碱性基体样品时,样品溶液中的钠离子和基体中和模块中的氢离子进行交换,乙醇钠和氢氧化钠分别变成乙醇和水,本实施例的中和仓模块再生液r2是草酸;
本实施例的基体中和模块是h离子交换柱,样品溶液s流经交换柱,溶液中的乙醇钠和氢氧化钠变为乙醇和水(钠离子被氢离子交换),再生液草酸是用来对h离子交换柱进行再生的(即草酸中的氢离子把交换柱上的钠离子洗脱下来)。
离子色谱仪分析金属钠中氯离子,使用包括淋洗液流路、再生液流路、进样流路和分析检测流路,所述分析检测流路由依次串联的阴离子交换柱、抑制器和电导检测池组成。样品溶液s通过进样流路进入离子色谱仪的基体中和模块和进样阀,样品溶液s中的钠离子和基体中和模块中的氢离子在此进行交换,乙醇钠和氢氧化钠分别变成乙醇和水;再经过基体消除装置,把溶液中的乙醇进行去除;在淋洗液e推动下,待测离子进入阴离子色谱交换柱,样品溶液s中的氯离子在阴离子交换柱内不断被吸附洗脱,使得氯离子与其他杂质分离,进入抑制器;在抑制器中,氯离子的信号值被增强,背景电导降低,样品溶液s最后进入电导检测器中检测生成谱图;
基体消除装置主要是预浓缩柱,载体是超纯水。具体流程如下:经过基体中和模块后,样品溶液s中主要成份有乙醇、氢离子、氯离子和其他阴离子,样品溶液s进入基体消除装置(基体消除装置主要为预浓缩柱),仪器中的超纯水作为载体带着样品溶液s流经预浓缩柱(预浓缩柱为阴离子交换柱),氯离子和其他阴离子被吸附至预浓缩柱上,乙醇、氢离子被超纯水冲走,排废,达到除去乙醇基体的目的。然后将预浓缩柱切换至离子色谱分析检测流路,用淋洗液e将预浓缩柱上氯离子和其他阴离子洗脱下来,实现进样。
在淋洗液e推动下,溶液进入阴离子色谱交换柱,这个阴离子色谱交换柱与基体消除装置的阴离子交换柱的区别是,这个交换柱比较长,能够使氯离子与其他各种杂质阴离子分离开。
在抑制器中,淋洗液e中的钠离子转换为氢离子,使氯离子的信号值增强,背景电导降低,随后进入电导检测器中检测生成谱图。抑制器为h离子交换柱,样品溶液s(包括碳酸钠、碳酸氢钠、氯离子和其他阴离子)进入抑制器后,溶液中各种阳离子(主要是钠离子)吸附到h型交换树脂上,与氢离子发生离子交换,氢离子进入溶液中,随后溶液进入电导检测器中检测生成谱图。在此过程中,氯化钠变成氯化氢,使得待测离子的电信号增强(阳离子中氢离子的电导信号最强);碳酸钠和碳酸氢钠变成碳酸分子,使得背景电导降低,因此提高了检测灵敏度。
s5.使用带有基体中和模块的离子色谱仪检测s2配制的空白溶液中的氯离子含量;
s6.采用标准加入法检验仪器准确度,采用步骤3配制的标准氯离子溶液,加入样品溶液s中,使用带有基体中和模块的离子色谱仪检测上述溶液中的氯离子含量;
s7.金属钠样品中的氯含量计算公式:(样品溶液s的氯离子浓度—空白溶液的氯离子浓度)*样品溶液s的体积/配制样品溶液s的金属钠质量。
本实施例所述淋洗液e为碳酸钠和碳酸氢钠的混合溶液,碳酸钠浓度为3.2mmol/l,碳酸氢钠浓度为1.0mmol/l。所述再生液r1为磷酸水溶液,溶液浓度为100mmol/l,所述再生液r2为草酸水溶液,溶液浓度为100mmol/l。
本实施例的技术方案采用了离子色谱法测量金属钠中的氯离子含量。离子色谱的工作原理是离子交换平衡,离子色谱中使用的固定相是离子交换树脂。离子交换树脂上分布有固定的带电荷的基团和能游动的配位离子。当样品加入离子交换色谱柱后,如果用适当的溶液洗脱,样品离子即与树脂上能游动的离子进行交换,并且连续进行可逆交换吸附和解吸,最后达到吸附平衡。采用本实施例的技术方案检测核级金属钠中的氯离子含量,具有以下优点:
(1)本技术方案使用带有基体中和模块的离子色谱检测核级金属钠中的微量氯离子含量,操作简单,减少了前处理时间,降低了操作危险性和繁琐的样品前处理带来的误差。
(2)本技术方案所述测试方法中,样品进行基体中和,大大降低了氯离子的检出限,同时,降低了离子交换色谱柱的消耗,节约了成本。
(3)本技术方案所述测试方法中,样品色谱图基线平稳,色谱峰形好,色谱峰可以完全分离,分析结果可信度高。
(4)本技术方案所述测试方法检出限低,金属钠中氯离子的方法检出限可达25ppb。
(5)本技术方案所述测试方法准确度高,加标回收率在92%~96%之间。
(6)本技术方案所述测试方法精确度高,相对标准偏差为8.33%(两块不同钠块,样品本身具有不均匀性)。
实施例2
本实施例在实施例1的基础上,结合具体实验数据对于本发明的技术方案进行进一步解释说明。
1、准备工作:
(1)试剂准备:
超纯水(18.2mω)
碳酸钠(优级纯)
碳酸氢钠(优级纯)
磷酸(85%,优级纯)
无水乙醇(优级纯)
草酸(99.8%,优级纯)
氯离子标准溶液(标准物质,1000ppm)
(2)氯离子标准溶液的配制
使用1000ppm的氯离子标准溶液,通过稀释法配制浓度为10ppb的氯离子标准溶液,仪器自动稀释为1ppb、2ppb、5ppb、10ppb。
(3)淋洗液的配制
使用碳酸钠(优级纯)和碳酸氢钠(优级纯)配制淋洗液,淋洗液浓度为3.2mmol/l的碳酸钠+1.0mmol/l碳酸氢钠。
(4)再生液的配制
使用磷酸(85%,优级纯)和草酸(99.8%,优级纯)配制抑制器再生液和基体中和模块再生液,抑制器再生液为100mmol/l磷酸溶液,基体中和模块再生液为100mmol/l草酸溶液。
(5)仪器配置
采用万通公司940离子色谱仪,仪器配置如表1所示。
表1仪器配制
(6)仪器参数:
表2仪器参数
2、操作步骤
(1)配制样品溶液
取4份约1.0g钠样品(内蒙古某钠厂,5#和8#钠块),放置在50ml聚乙烯试管中,贴好标签,分别为5#1、5#2、8#1、8#2,取30ml无水乙醇溶解20分钟。刚开始反应剧烈有大量气泡产生,需在通风柜中进行。
溶解20分钟后,反应减缓,可将样品溶液放置在石墨电加热板上70℃加热15分钟,加热完毕如仍残留少量钠块,可逐滴加入1-2ml超纯水加速溶解。待完全溶解后,加超纯水定容至50ml,得到5#1、5#2、8#1、8#2样品溶液。
(2)配制空白溶液
在50ml聚乙烯试管中加入30ml无水乙醇与上述样品溶液进行同样操作,最后同样定容至50ml,摇匀备用。
(3)配制氯离子标准溶液
将1000ppm氯离子标准溶液,加入超纯水,稀释,配制成10ppb浓度标准溶液。另再配制1ppm氯离子标准溶液,摇匀备用。
(4)标准曲线的绘制
取10ppb氯离子标准溶液进仪器,仪器自动稀释为1.0ppb、2.0ppb、5.0ppb、10.0ppb浓度,也经过相同流路被检测,输出谱图,并拉直标准曲线。从图8、图9可以看出:所获工作曲线线性关系良好,相关系数大于0.999。
表3仪器自动稀释标准氯离子溶液浓度
(5)空白溶液检测
准确移取1ml空白溶液,加水9ml,稀释10倍后,进样,连续进样7次。在淋洗液e的推动下,空白溶液先进过基体中和模块和进样阀,进入阴离子色谱柱,氯离子在阴离子交换柱中被吸附和洗脱,达到分离后进入抑制器,在抑制器中,将淋洗液中的碱中和成水,降低背景电导值后进入电导检测池被检测,空白溶液被检测,以谱图形式输出。测量结果如下表4,测量谱图如图1、图2、图3所示。
表4空白测量结果
由表4可知,将空白溶液进行上述前处理方式处理后,进入仪器检测,溶液中氯离子的方法检出限为0.051ppb。
(6)样品溶液检测
准确移取5#1、5#2、8#1、8#2样品溶液各1ml,加水9ml,稀释10倍后,进样。试样溶液也经过相同的流路被检测,输出谱图,如图4、图5、图6、图7所示。测量结果如表5:
表5试样溶液测量结果
由表5可知,4个样品的相对标准偏差为8.33%,满足示范快堆核级金属钠中氯分析的精确度要求。
(7)样品溶液加标后检测
准确移取1ml5#1和8#2试样溶液,分别加入0.04ml,0.08ml和0.16ml的1mg/l的氯离子标准溶液,定容到10ml,此时加标量分别为4ppb,8ppb和16ppb。将加标后样品溶液重复步骤(4),计算回收率。图10、图11为加标后测量谱图。表6为加标后测量结果。
表6样品加标回收结果
由表6可知,样品的加标回收率在92%~96%之间,满足示范快堆核级金属钠中氯分析的准确度要求。
(8)金属钠样品中的氯离子含量计算
将试样5#1、5#2、8#1、8#2溶液测量值减去空白值(7次空白测量的平均值),即得到4份试样中氯离子含量,测量结果见下表7
表7试样中氯离子含量
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。倘若这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
- 还没有人留言评论。精彩留言会获得点赞!