金水六君煎指纹图谱的建立方法与流程
本发明涉及中药检测技术领域,尤其涉及金水六君煎指纹图谱的建立方法。
背景技术:
金水六君煎主要用于肺肾虚寒,水泛为痰症,咳嗽呕恶等症,由当归、熟地黄、陈皮、半夏茯苓及炙甘草六味药组成,现代临床主要用于以咳、喘为主要临床表现的疾病,具有丰富的临床疗效基础,是国家中医药管理局发布的《古代经典名方目录(第一批)》所载方剂之一。
目前,中药的质量标准难以统一,同一味中药治疗不同疾病时主要有效成分不同,经典名方要求处方药材质量均一、稳定,故研发前确定各味中药的基原,对处方中各味药材的临床用药情况、药材来源变化和历版《中国药典》的修订情况进行深入的考证确定具有非常重要的意义。现今中药质量标准主要是对有效成分以及有效部位含量的测定,但中药成分较为复杂,单一有效成分或有效部位难以阐明中药质量,中药组合物的质量标准怎么制定以及能否制定出较为公认的评价标准是大家关注的焦点。
因此,为了更好的反应经典名方金水六君煎的药用质量,建立金水六君煎指纹图谱是非常必要的。
技术实现要素:
针对现有金水六君煎的质量和评价标准无法统一的问题,本发明提供一种金水六君煎指纹图谱的建立方法。
为达到上述发明目的,本发明实施例采用了如下的技术方案:
金水六君煎指纹图谱的建立方法,包括以下步骤:
a、供试品溶液的制备:按金水六君煎中各组分的质量配比分别称取熟地黄、当归、炙甘草、茯苓、半夏和陈皮,加水和姜片浸泡,煎煮,过滤得到滤液,即为供试品溶液;
b、对照品溶液的制备:用溶剂溶解橙皮苷和甘草酸铵,得到混合溶液,即为对照品溶液;
c、高效液相色谱检测:将供试品溶液和对照品溶液分别用高效液相色谱进行检测,得到金水六君煎指纹图谱,所述高效液相色谱条件为:
固定相:c18色谱柱;
流动相:流动相a相为乙腈,b相为0.1-0.3wt%的磷酸溶液;
梯度洗脱条件为:0-10min,a相5-8vol%,b相95-92vol%;10-35min,a相8-14vol%,b相92-86vol%;35-50min,a相14-16vol%,b相86-84vol%;50-65min,a相16-21vol%,b相84-79vol%;65-85min,a相21-32vol%,b相79-68vol%;85-95min,a相32-40vol%,b相68-60vol%;95-120min,a相40-65vol%,b相60-35vol%;120-125min,a相65-5vol%,b相35-95vol%;125-130min,a相5vol%,b相95vol%。
相对于现有技术,本发明提供的金水六君煎有效成分指纹图谱的建立方法,得到的金水六君煎指纹图谱与对照指纹图谱的相似度大于0.9以上,稳定性好,可重复性好,不同批次间差异小,且同批次之间均一性好、稳定性高,得到的指纹图谱信息量大、谱峰信息丰富,共有峰可达到12个,且每个共有峰分别为金水六君煎中的一种有效药物成分,可非常全面的提供金水六君煎的整体药用成分信息,不仅有助于为金水六君煎制定相关具体的质量标准,而且为后期金水六君煎成分的鉴定以及挖掘药效物质奠定了基础。
本发明固定相采用c18色谱柱,流动相的a相为乙腈,b相为0.1-0.3wt%的磷酸溶液,该固定相和流动相的结合,配合上述特定的洗脱条件可以增加金水六君煎提取液中各有效成分的分离度,可有效提高指纹图谱共有峰的数量,增加金水六君煎指纹图谱的信息量,且共有峰的峰形、分离度较好,色谱峰响应好,即使以水作为提取溶剂并将提取液直接进行高效液相色谱,也能达到上述色谱效果,免去有机提取溶剂和有机提取溶媒等有机溶剂的加入,保证了指纹图谱检测的通用性和有效性。
本发明建立金水六君煎指纹图谱的方法可对物质基准中的某些化学成分进行定性分析,对评价物质基准的质量提供重要依据。
优选的,所述步骤a中水的加入量为金水六君煎质量的7-8倍,浸泡时间为20-30min;煎煮过程为:武火煎煮8-10min,文火煎煮20-22min。
本发明的金水六君煎有效成分指纹图谱的建立方法既保证了古法煎煮工艺,以原始的水煎液注入液相进行分析,同时对古法煎煮工艺进行改进,通过设置特定的煎煮方式和煎煮时间,可使药物煎煮至最佳状态,使药物的浸膏得率达到最大。
优选的,所述步骤b中的溶剂为80-100vol%的甲醇溶液。
优选的,所述高效液相色谱的固定相采用依利特c18色谱柱;所述高效液相色谱的流动相a相为乙腈,b相为0.1wt%的磷酸溶液。
采用依利特c18色谱柱,配合乙腈-0.1wt%的磷酸溶液作为流动相,建立的指纹图谱的共有峰之间的分离度最高,峰形最好。
优选的,所述步骤c中高效液相色谱的流动相流速为0.7-0.8ml/min,色谱柱柱温为34-36℃,检测波长为260-270nm。
上述色谱柱柱温和流动相流速的设置可以增加色谱图中相应成分的分离程度。
优选的,所述流动相的流速为0.8ml/min。
0.8ml/min的流速,刚好满足色谱柱对金水六君煎中各成分的分离速度。
优选的,所述高效液相色谱的色谱柱柱温为35℃,检测波长为260nm。
260nm为金水六君煎指纹图谱建立的最佳吸收波长,该波长条件下,各共有峰的基线平稳且响应良好。
附图说明
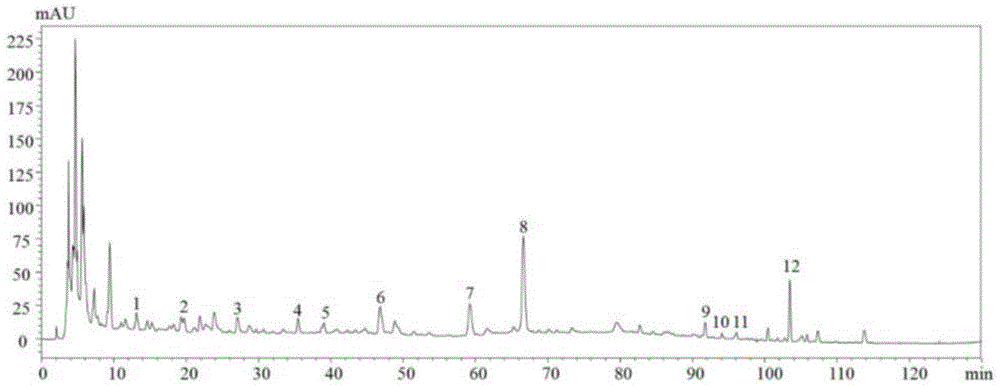
图1是本发明实施例中金水六君煎供试品溶液的高效液相色谱图;
图2是本发明实施例中对照品溶液的高效液相色谱图;
图3是本发明实施例中17批金水六君煎的指纹图谱;其中r为共有模式;
图4是本发明实施例中17批金水六君煎指纹图谱聚类树状关系图;
图5是本发明实施例中17批金水六君煎指纹图谱碎石图;
图6是本发明实施例中根据17批金水六君煎指纹图谱中12个共有峰峰面积进行的主成分分析图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
实施例1
金水六君煎指纹图谱的建立方法
1、仪器与试药
仪器:日本岛津lc-20at高效液相色谱仪,spd-20紫外检测器,岛津labsolution工作站,auw220d型电子天平(十万分之一,日本岛津公司);bs224s型电子天平(万分之一,sartorius);kq250db型数控超声波清洗器(巩义市予华仪器有限责任公司);kq5200型水浴锅(昆山市超声仪器有限公司);
试药:乙腈(色谱纯)(上海振兴化工一厂),磷酸(广东汕头市西陇化工有限公司),橙皮苷对照品(批号:0856-200203,供含量测定用),甘草酸铵对照品(批号:0856-200203,供含量测定用)(中国食品药品检定研究院);收集的处方药味信息如表1所示:
表1处方药味信息
将收集到的处方药味信息随机组合成17批金水六君煎,如表2所示:
表2随机组合的17批金水六君煎
2、方法与结果
供试品溶液制备:取熟地黄15g,当归7.5g,炙甘草3.75g,茯苓7.5g,半夏7.5g,陈皮7.5g,生姜3片,加水400ml浸泡20min,武火煎煮9分钟,文火煎煮21分钟,滤过,即得;
对照品溶液制备:分别取橙皮苷5.45mg、甘草酸铵3.75mg,精密称定,置于25ml量瓶中,用100vol%甲醇溶解并稀释,制成每1ml含橙皮苷0.218mg、甘草酸铵0.15mg的混合溶液;
高效液相色谱检测:采用岛津lc-20at型高效液相色谱仪,固定相为依利特c18色谱柱(4.6×250mm,4.6μm),流动相为乙腈(a)-0.1%磷酸(b);梯度洗脱条件为:0-10min,a相5-8vol%,b相95-92vol%;10-35min,a相8-14vol%,b相92-86vol%;35-50min,a相14-16vol%,b相86-84vol%;50-65min,a相16-21vol%,b相84-79vol%;65-85min,a相21-32vol%,b相79-68vol%;85-95min,a相32-40vol%,b相68-60vol%;95-120min,a相40-65vol%,b相60-35vol%;120-125min,a相65-5vol%,b相35-95vol%;125-130min,a相5vol%,b相95vol%;柱温35℃,流速:0.8ml/min,波长260nm。
高效液相色谱得到的供试品溶液的色谱图如图1所示,对照品溶液的色谱图如图2所示。
3、方法学考察
精密度:取供试品溶液,按照上述色谱条件连续进样6次,记录色谱图。以12号色谱峰(甘草酸铵)为参照峰,计算共有峰相对保留时间、相对峰面积的rsd均低于5.0%,表明仪器精密度良好。
重复性:取同批供试品溶液6份,按上述色谱条件测定,记录色谱图,以12号色谱峰(甘草酸铵)为参照峰,计算共有峰相对保留时间、相对峰面积的rsd均低于5.0%,表明该方法重复性良好。
稳定性:取供试品溶液1份,分别在制备后0,2,4,8,12,24h按上述色谱条件测定,记录色谱图,以12号色谱峰(甘草酸铵)为参照峰,计算共有峰相对保留时间、相对峰面积的rsd均低于5.0%,表明物质基准供试品溶液在24h内稳定性良好。
4、指纹图谱的建立和检测分析
指纹图谱的建立:将随机组合的17批金水六君煎分别按照上述方法得到色谱图,并将色谱图数据导入“中药色谱指纹图谱相似度评价系统”(2012版)软件,设置参照图谱,并标记12个共有峰,生成指纹图谱,如图3所示。
相似度评价:17批金水六君煎指纹图谱与其生成的对照指纹图谱的相似度均大于0.9,如表3所示,说明制备工艺稳定,物质群批间差异小。
表317批金水六君煎指纹图谱相似度
数据分析:
spss分析:将17批金水六君煎的12个共有峰面积导入ibmspssstatistics软件进行组间聚类分析,其中样品间距离采用euclidean距离法进行计算,得到17批金水六君煎指纹图谱聚类树状关系图(如图4所示)和碎石图(如图5所示),通过图4的聚类分析表明,17批样品可分为2类,其中wzjz-1、2、3、4、5、10及13可聚为一类,其余批次样品可聚为一类;
对12个标记峰峰面积以主成分的特征值及贡献率为依据,进行主成分因子分析,计算相关系数矩阵,主成分特征值、累积贡献率并计算主成分综合得分,对所制备的17批金水六君煎进行评价,结果见表5,由表4和图5可知,12个标记成分中,峰1、2、3的特征值较大(>1)且连线较陡峭,累积方差贡献率89.495%,以因子的方差累计贡献率85%以上作为主成分提取标准,提取前3个成分可整体反映指标89.495%以上的信息,其中提取主成分1的特征值为8.319,贡献率为69.324%;提取主成分2的特征值为1.418,贡献率为11.820%;提取主成分3的特征值为1.002,贡献率为8.351%。
表4金水六君煎总方差解释变异量
simca分析:记录的17批金水六君煎的12个共有峰峰面积,运用simca13.0软件进行主成分分析,得到图6,从图6的pca图可以看出,wzjz-1、2、3、4、5、10及13可聚为一类,其余批次样品可聚为一类。
综合上述分析结果表明:
17批金水六君煎指纹图谱标记的12个共有峰的相似度在0.9以上,说明不同批次间差异性不大,而12个共有峰峰面积经spss和simca分析后,发现同法煎煮的不同批次物质基准存在一定的差异,17批物质基准可分为2类。
标记峰1、2、3为主要成分的影响因素,金水六君煎中各组分是收集不同批次不同产地的处方药材随机组合而得,不同的药材进行混合煎煮,对某些成分产生的助溶或抑溶作用会对某些成分含量会产生差异性,因此,在综合评价时,指纹图谱的相似度不足以定论其优劣,通过主成分、因子和聚类分析,相互验证和补充,多方面评价物质基准的质量,对多角度评价物质基准具有重要意义,经典名方金水六君煎目前无相关的药效筛选研究,本发明所建立的指纹图谱方法不仅有助于为金水六君煎物质基准序制定相关具体的质量标准,而且为后期利用液质联用等技术对成分的鉴定提供参考,从而挖掘金水六君煎物质基准的药效物质奠定了基础。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换或改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!