一种康肾颗粒指纹图谱的构建方法及其标准指纹图谱与流程
本发明属于中药制剂质量控制技术领域,更具体地,涉及一种康肾颗粒指纹图谱的构建方法及其标准指纹图谱。
背景技术:
我国传统中药复杂,化学成分复杂,其药效作用往往难以用分单一成分表达。我国现有中药及其制剂质量标准相对较为简单,大多局限于外观性状、鉴别,部分品种建立化学成分含量测定方法,即使在已经建立化学成分测定药材的方法中,其所测定成分也存在专属性不强,与药效没有直接关系,难以对药材和制剂的有效性与安全性进行全面的描述。对中药的内在质量难以进行有效的控制,无法保证用药的安全、有效,严重制约了我国中药行业的发展。为加强对中药的质量控制,促进中药规范化、现代化和标准化,采用先进的科学方法对中药进行有效的质量控制研究意义重大。
康肾颗粒由云南彝族经验方转化的制剂,处方是由连钱草、忍冬藤、石韦、白茅根、石菖蒲、葛根、茜草、艾叶、生姜、陈皮、水蜈蚣、老鹳草12味药材组成,具有补脾益肾,化湿降浊的作用,临床上具有显著的效果。康肾颗粒收载于国家食品药品监督管理局国家药品标准中,其现有质量标准较为简单,无法对康肾颗粒进行全面的质量控制。
针对康肾颗粒,目前缺乏全面反映其质量和真伪的方法,且目前并未有关于康肾颗粒指纹图谱的研究报道,因而,构建针对康肾颗粒指纹图谱的研究,对产品质量控制具有较大的意义,能够为康肾颗粒提供一种全面的质量控制方法。
技术实现要素:
本发明的目的在于根据现有技术中的不足,提供了一种康肾颗粒指纹图谱的构建方法。
本发明的另一目的在于提供上述构建方法建立的标准指纹图谱。
本发明的目的通过以下技术方案实现:
本发明提供了一种康肾颗粒指纹图谱的构建方法,包括如下步骤:供试品溶液和对照品溶液的制备,高效液相色谱条件的确定,指纹图谱的确定;
所述供试品溶液的制备为取康肾颗粒样品0.5~5.0重量份,加入50%~100%的甲醇15~50体积份,提取15~60分钟,过滤,取续滤液,即得;
所述高效液相色谱条件为以十八烷基硅烷键合硅胶为填充剂;检测波长为270~290nm;柱温为20~40℃;流速为0.8~1.2ml/min;流动相由流动相A和B组成,以乙腈为流动相A,以0.5~1.5%的甲酸水溶液为流动相B;进行线性梯度洗脱;
线性梯度洗脱条件为:
从0~35min,流动相A的体积分数为5~12%;流动相B的体积分数为95~88%;
从35~60min,流动相A的体积分数为12~20%;流动相B的体积分数为88~80%;
从60~85min,流动相A的体积分数为20~40%;流动相B的体积分数为80%~60%;
从85~100min,流动相A的体积分数为40~90%;流动相B的体积分数为60~10%;
从100~110min,流动相A的体积分数为90~95%;流动相B的体积分数为10~5%。
优选地,所述提取的方法为超声提取法或加热回流法。
优选地,所述供试品制备为取康肾颗粒样品2.0重量份,加入75%的甲醇25体积份,提取30分钟,过滤,取续滤液,即得。所述高效液相色谱条件中检测波长为280nm;柱温为25℃;流速为1.0ml/min;流动相由流动相A和B组成,以乙腈为流动相A,以1.0%的甲酸水溶液为流动相B。
优选地,所述的供试品制备方法为取康肾颗粒样品5.0重量份,加入100%的甲醇50体积份,提取60分钟。所述高效液相色谱条件中检测波长为290nm;柱温为40℃;流速为1.2ml/min;流动相由流动相A和B组成,以乙腈为流动相A,以1.5%的甲酸水溶液为流动相B。
优选地,所述的供试品制备方法为取康肾颗粒样品0.5重量份,加入50%的甲醇15体积份,提取15分钟。所述高效液相色谱条件为中检测波长为270nm;柱温为20℃;流速为0.8ml/min;流动相由流动相A和B组成,以乙腈为流动相A,以0.5%的甲酸水溶液为流动相B。
优选地,所述对照品溶液的制备为:分别取葛根素、咖啡酸、大豆苷、迷迭香酸、橙皮苷、大豆苷元对照品,加入甲醇,配制成浓度为葛根素90.0μg/ml、咖啡酸52.6μg/ml、大豆苷127.2μg/ml、迷迭香酸80.4μg/ml、橙皮苷19.1μg/ml、大豆苷元42.8μg/ml的对照品溶液。
优选地,高效液相色谱条件中进样量为10μL。
本发明中提及的重量份与体积份的关系为g/ml的关系;
中药指纹图谱技术是一种控制中药质量的现代化关键技术,能全面的反应药物所含化学成分的种类和数量,对药物的物质基础进行全面的阐释,进而更好的控制药物内在质量。
而在中药指纹图谱的构建方法中,为了使指纹图谱中各特征峰具有较好的分离度、峰形等,色谱条件(柱温、检测波长、流动相种类、梯度洗脱程序、流速等)和供试品制备方法(制备方式、制备时间、制备溶液等)对指纹图谱影响较大。且由于对于不同的中药材及制剂,其中的化学成为千差万别,不同的药材及制剂的指纹图谱的构建方法并无参考价值。因此申请人在大量实验基础上,针对康肾颗粒中各种成分的特性,进行色谱条件以及供试品制备方法进行了摸索及优化,最终确定上述条件,且获得了显著的分离效果。
本发明另外提供一种根据所述的构建方法得到的康肾颗粒标准指纹图谱,所述标准指纹图谱含有27个共有峰,以葛根素峰为参照,27个共有峰相对保留时间比依次为:0.171~0.173,0.245~0.247,0.291~0.293,0.362~0.365,0.520~0.524,0.693~0.696,0.740~0.742,0.806~0.808,0.849~0.853,1.000,1.022~1.023,1.078~1.080,1.099~1.101,1.152~1.154,1.259~1.261,1.287~1.290,1.4996~1.502,1.545~1.550,1.758~1.766,1.889~1.900,1.979~1.990,2.086~2.098,2.121~2.137,2.168~2.182,2.181~2.196,2.349~2.367,2.700~2.719;
优选地,本发明另外提供一种根据所述的构建方法得到的康肾颗粒标准指纹图谱,所述标准指纹图谱含有27个共有峰,以葛根素峰为参照,27个共有峰相对保留时间比依次为:0.172,0.246,0.292,0.364,0.523,0.695,0.741,0.807,0.852,1.000,1.023,1.079,1.100,1.153,1.260,1.288,1.499,1.547,1.763,1.895,1.986,2.094,2.129,2.176,2.189,2.358,2.710。
本发明提供的方法,其精密度、重复性和稳定性实验中,共有峰的相对保留时间的RSD均小于5%,相对峰面积的RSD均小于5%,相似度均大于0.900,样品在48个小时内稳定性良好。
针对该方法所获得康肾颗粒指纹图谱,能全面的反应药物所含化学成分的种类和数量,对药物的物质基础进行全面的阐释,能够准确地对多批次样品进行相似度分析,进而更好的控制药物内在质量。
与现有技术相比,本发明具有以下优点及有益效果:
本发明所构建的康肾颗粒指纹图谱,有多个共有峰的色谱信息构成,改变单一指标成分进行质量控制的技术手段,能够系统全面地反映其物质基础,从而进行全面、有效的质量控制,并且所建立的方法精密度、重复性、稳定性较好,可用于康肾颗粒内在质量的控制。
附图说明
图1为精密度试验指纹图谱叠加图。
图2为重复性试验指纹图谱叠加图。
图3为稳定性试验指纹图谱叠加图。
图4为康肾颗粒13批样品指纹图谱叠加图。
图5为混合对照品指纹图谱。
图6为对照指纹图谱。
图7为药材与样品指纹图谱匹配叠加图。
具体实施方式
以下结合具体实施例和附图来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
除非特别说明,本发明所用试剂和材料均为市购。
仪器、试剂及样品信息如下:
1、仪器
2695-2998型waters高效液相色谱仪(waters公司),CPA225D型电子分析天平(d=0.01mg,赛多利斯科学仪器(北京)有限公司),SB-5200D型超声仪(宁波新芝生物科技股份有限公司)、HWS24型恒温水浴锅(上海一恒科学仪器有限公司)、Waters Summetry C18色谱柱(4.6×250mm,5μm)(waters公司)。
2、试剂
对照品:葛根素对照品(110752-201107,中国食品药品检定研究院)、大豆苷元对照品(111502-200402,中国食品药品检定研究院)、大豆苷对照品(111738-201302,中国食品药品检定研究院)、橙皮苷对照品(110721-201316中国食品药品检定研究院)、迷迭香酸对照品(111871-2014,中国食品药品检定研究院)、咖啡酸对照品(110885-200102,中国食品药品检定研究院)
色谱甲醇购自OCEANPAK公司,色谱乙腈购自Burdick&Jackson公司,甲醇、无水乙醇、甲酸等均为色谱纯,水为自制超纯水。
3、样品
单味药材连钱草、忍冬藤、石韦、白茅根、石菖蒲、葛根、茜草、艾叶、生姜、陈皮、水蜈蚣、老鹳草经广州一品红制药有限公司质量部鉴定为正品,其中连钱草为唇形科植物活血丹Glechoma longituba(Nakai)Kupr.的干燥地上地上部分,忍冬藤为忍冬科植物忍冬Lonicera japonica Thunb.的干燥茎枝,石韦为水龙骨科植物庐山石韦Pyrrasia sheareri(Bak.)Ching、石韦Pyrrosia lingua(Thunb.)Farwell或有柄石韦Pyrrosia petiolosa(Christ)Ching的干燥叶、白茅根为禾本科植物白茅Imperata cylindrical Beauv.var.major(Nees)C.E.Hubb.的干燥根茎,石菖蒲为天南星科植物石菖蒲Acorus tatarinowii Schott的干燥根茎,葛根为豆科植物野葛Pueraria lobata(Willd.)Ohwid的干燥根,茜草为茜草科植物茜草Rubia cordifolia L.的干燥根和根茎,艾叶为菊科植物艾Artemisia argyi Levl.et Vant.的干燥叶,生姜为姜科植物姜Zingiber officinale Rosc.的新鲜根茎,陈皮为芸香科植物橘Citrus reticulat Blanco及其栽培变种的干燥成熟果实,水蜈蚣为莎草科植物水蜈蚣Kyllinga brevifolia Rottb.的干燥全草,老鹳草为牻牛儿苗植物牻牛儿苗Erodium stephanianum Wild.、老鹳草Geranium wilordii Maxim.或野老鹳草Geranium carolinianum L.的干燥地上部分。
康肾颗粒(规格:12g/袋)13批,编号及批号见表1
表1样品信息表
实施例1
1、供试品溶液的制备:取样品约2.0g,精密称定,置具塞锥形瓶中,精密加入75%甲醇25ml,称定重量,超声提取法提取30分钟,放冷,再称定重量,用75%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2、对照品溶液的制备:取葛根素、咖啡酸、大豆苷、迷迭香酸、橙皮苷、大豆苷元对照品,精密称定,加甲醇配制成浓度为葛根素90.0μg/ml、咖啡酸52.6μg/ml、大豆苷127.2μg/ml、迷迭香酸80.4μg/ml、橙皮苷19.1μg/ml、大豆苷元42.8μg/ml的混合对照品溶液。
3、高效液相色谱条件:色谱柱以十八烷基硅烷键合硅胶为填充剂;检测波长为280nm;柱温为25℃;以乙腈为流动相A,以1.0%甲酸水为流动相B;梯度洗脱;流速为1.0ml/min;梯度洗脱程序为:
0~35分钟,流动相A从5%变为12%,流动相B为95%变为88%;
35~60分钟,流动相A从12%变为20%,流动相B为88%变为80%;
60~85分钟,流动相A从20%变为40%,流动相B为80%变为60%;
85~100分钟,流动相A从40%变为90%,流动相B为60%变为10%;
100~110分钟,流动相A从90%变为95%,流动相B为95%变为5%;
样品测定法,分别精密吸取供试品溶液与对照品溶液10μl,注入高效液相色谱仪测定,即得指纹图谱。
所构建康肾颗粒指纹图谱,共有27个共有峰,与对照品指纹图谱对照出峰时间对比,按出峰顺序确定8号峰为咖啡酸、10号峰为葛根素、15号峰为大豆苷、20号峰为橙皮苷、21号峰为迷迭香酸、22号峰为大豆苷元。以10号峰葛根素为参照峰,27个共有峰相对保留时间比依次为:0.172,0.246,0.292,0.364,0.523,0.695,0.741,0.807,0.852,1.000,1.023,1.079,1.100,1.153,1.260,1.288,1.499,1.547,1.763,1.895,1.986,2.094,2.129,2.176,2.189,2.358,2.710。
实施例2
1、供试品溶液的制备:取本品约0.5g,精密称定,置具塞锥形瓶中,精密加入50%甲醇15ml,称定重量,超声提取法提取15分钟,放冷,再称定重量,用50%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2、对照品溶液的制备:取葛根素、咖啡酸、大豆苷、迷迭香酸、橙皮苷、大豆苷元对照品,精密称定,加甲醇配制成浓度为葛根素90.0μg/ml、咖啡酸52.6μg/ml、大豆苷127.2μg/ml、迷迭香酸80.4μg/ml、橙皮苷19.1μg/ml、大豆苷元42.8μg/ml的混合对照品溶液。
3、高效液相色谱条件:色谱柱以十八烷基硅烷键合硅胶为填充剂;检测波长为270nm;柱温为20℃;以乙腈为流动相A,以0.5%甲酸水为流动相B;梯度洗脱;流速为0.8ml/min;梯度洗过程序为:
0~35分钟,流动相A从5%变为12%,流动相B为95%变为88%;
35~60分钟,流动相A从12%变为20%,流动相B为88%变为80%;
60~85分钟,流动相A从20%变为40%,流动相B为80%变为60%;
85~100分钟,流动相A从40%变为90%,流动相B为60%变为10%;
100~110分钟,流动相A从90%变为95%,流动相B为95%变为5%;
样品测定法,分别精密吸取供试品溶液与对照品溶液10μl,注入高效液相色谱仪测定,即得指纹图谱。
所构建康肾颗粒指纹图谱,共有27个共有峰,与对照品指纹图谱对照出峰时间对比,按出峰顺序确定8号峰为咖啡酸、10号峰为葛根素、15号峰为大豆苷、20号峰为橙皮苷、21号峰为迷迭香酸、22号峰为大豆苷元。以10号峰葛根素为参照峰,27个共有峰相对保留时间比依次为:0.171,0.245,0.291,0.362,0.520,0.693,0.740,0.806,0.849,1.000,1.022,1.078,1.099,1.152,1.259,1.287,1.4996,1.545,1.758,1.889,1.979,2.086,2.121,2.168,2.181,2.349,2.700。
实施例3
1、供试品溶液的制备:取本品约5.0g,精密称定,置具塞锥形瓶中,精密加入100%甲醇50ml,称定重量,加热回流法提取60分钟,放冷,再称定重量,用100%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2、对照品溶液的制备:取葛根素、咖啡酸、大豆苷、迷迭香酸、橙皮苷、大豆苷元对照品,精密称定,加甲醇配制成浓度为葛根素90.0μg/ml、咖啡酸52.6μg/ml、大豆苷127.2μg/ml、迷迭香酸80.4μg/ml、橙皮苷19.1μg/ml、大豆苷元42.8μg/ml的混合对照品溶液。
3、高效液相色谱条件:色谱柱以十八烷基硅烷键合硅胶为填充剂;检测波长为290nm;柱温为40℃;以乙腈为流动相A,以1.5%甲酸水为流动相B;梯度洗脱;流速为1.2ml/min;梯度洗过程序为:
0~35分钟,流动相A从5%变为12%,流动相B为95%变为88%;
35~60分钟,流动相A从12%变为20%,流动相B为88%变为80%;
60~85分钟,流动相A从20%变为40%,流动相B为80%变为60%;
85~100分钟,流动相A从40%变为90%,流动相B为60%变为10%;
100~110分钟,流动相A从90%变为95%,流动相B为95%变为5%;
样品测定法,分别精密吸取供试品溶液与对照品溶液10μl,注入高效液相色谱仪测定,即得指纹图谱。
所构建康肾颗粒指纹图谱,共有27个共有峰,与对照品指纹图谱对照出峰时间对比,按出峰顺序确定8号峰为咖啡酸、10号峰为葛根素、15号峰为大豆苷、20号峰为橙皮苷、21号峰为迷迭香酸、22号峰为大豆苷元。以10号峰葛根素为参照峰,27个共有峰相对保留时间比依次为:0.173,0.247,0.293,0.365,0.520~0.524,0.696,0.742,0.808,0.853,1.000,1.023,1.080,1.101,1.154,1.261,1.290,1.502,1.550,1.766,1.900,1.990,2.098,2.137,2.182,2.196,2.367,2.719。
实施例4:
1、精密度试验
取批号为20151201批次样品,按实施例1方法制备供试品溶液,依据实施例1色谱条件连续进样6次,以相对保留时间、相对峰面积、相似度为考察指标,结果显示,共有峰的相对保留时间的RSD均小于5%,相对峰面积的RSD均小于5%,相似度均大于0.900,符合指纹图谱的要求,表明仪器的精密度良好。
所得指纹图谱叠加图见图1。
同时,分别按照实施例2、3中条件进行上述精密度实验,共有峰的相对保留时间的RSD均小于5%,相对峰面积的RSD均小于5%,相似度均大于0.900,表明采用实施例2、3中的方法也能符合指纹图谱的要求。
2、重复性试验
取批号为20151201批次样品6份,按实施例1方法制备供试品溶液,依据实施例1色谱条件连续进样6次,以相对保留时间、相对峰面积、相似度为考察指标,结果显示共有峰的相对保留时间的RSD均小于5%,相对峰面积的RSD均小于5%,相似度均大于0.900,符合指纹图谱的要求,表明方法的重复性良好。
所得指纹图谱叠加图见图2。
同时,分别按照实施例2、3中条件进行上述重复性实验,与共有峰的相对保留时间的RSD均小于5%,相对峰面积的RSD均小于5%,相似度均大于0.900,表明采用实施例2、3中的方法也能符合指纹图谱的要求。
3、稳定性试验
取精密度试验样品,依据实施例1下色谱条件分别在0、6、10、16、26、39、48小时进样,以相对保留时间、相对峰面积、相似度为考察指标,结果显示共有峰的相对保留时间的RSD均小于5%,相对峰面积的RSD均小于5%,相似度均大于0.900,符合指纹图谱的要求,表明样品在48个小时内稳定性良好。
所得指纹图谱叠加图见图3。
同时,分别按照实施例2、3中条件进行上述稳定性实验,共有峰的相对保留时间的RSD均小于5%,相对峰面积的RSD均小于5%,相似度均大于0.900,表明采用实施例2、3中的方法也能符合指纹图谱的要求。
实施例5:
1、按照实施例1条件,取康肾颗粒样品溶液、混合对照品溶液,药材供试品溶液(药材供试品溶液制备:分别取处方中12味药材,每份取约1g,精密称定,按照实施例1中方法供试品制备方法制备药材供试品溶液。),依据实施例1下色谱条件进样,记录色谱图。
2、对照指纹图谱的建立
对13批样品指纹图谱进行数据处理:
导入国家药典委员会的《中药色谱指纹图谱相似度评价系统》,平均值法生成对照指纹图谱,时间窗宽度为0.1min,校正方式为多点校正。
确定了27个色谱峰为康肾颗粒共有指纹峰,结合供试品以及对照品的保留时间以及DAD色谱图,可知共有峰中8号峰为咖啡酸、10号峰为葛根素、15号峰为大豆苷、20号峰为橙皮苷、21号峰为迷迭香酸、22号峰为大豆苷元。
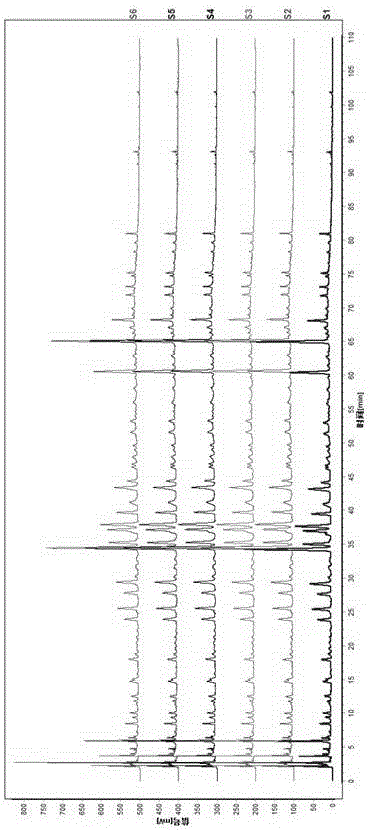
13批样品指纹图谱、混合对照品指纹图谱、对照指纹图谱分别见图4~6。
从图4中也可以看出,利用该方法检测到的康肾颗粒指纹图谱中化学成分多、各特征峰高度适中,基线平稳,分离度、峰形和柱效好。
实施例6:
采用国家药典委员会的《中药色谱指纹图谱相似度评价系统》,将测定的13批样品指纹图谱数据与生成的对照图谱进行相似度分析,13批样品的相似度见表2,从表中数据来看,13批样品相似度均在0.980以上,说明本品的质量稳定性较好。
表2 13批样品相似度
实验例7:色谱峰归属
取康肾颗粒供试品溶液、混合对照品溶液、药材供试品溶液(同实施例5)。按实施例1中色谱条件进样,记录色谱图。通过色谱柱匹配和分析,对共有峰进行归属,见表3和图7。
其中,有10个成分归属到连钱草、7个成分归属到忍冬藤,4个成分归属到石韦,7个成分归属到陈皮,4个成分归属到白茅根,6个成分归属到石菖蒲,10个成分归属到葛根,6个成分归属到茜草,9个成分归属到艾叶,1个成分归属到生姜,5个成分归属到水蜈蚣,3个成分归属到老鹳草,其中24号色谱峰没有药材归属。
据此,得到康肾颗粒指纹图谱,所述指纹图谱含有27个共有峰,以葛根素峰为参照,27个共有峰相对保留时间比依次为:0.172,0.246,0.292,0.364,0.523,0.695,0.741,0.807,0.852,1.000,1.023,1.079,1.100,1.153,1.260,1.288,1.499,1.547,1.763,1.895,1.986,2.094,2.129,2.176,2.189,2.358,2.710。
表3指纹图谱共有峰归属
可见,本发明所述康肾颗粒指纹图谱的构建方法,可全面反映康肾颗粒的物质基础信息,且方法准确可行。
实施例8
分别取康肾颗粒成品三批,批号分别为20160101、20160204、20160304,按照实施例1的方法操作得到样品指纹图谱,导入《中药色谱指纹图谱相似度评价系统》,采用“分析检验”模式,与实施例7中康肾颗粒对照指纹图谱进行比对,计算其相似度,结果三批样品相似度分别为0.951、0.965、0.950,说明三批康肾颗粒物质基础与对照指纹图谱一致。
显然,上述实施例仅仅是为清楚的说明所做的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以作出不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申的显而意见的变化或变动仍处于本发明创造的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!