一种基于结构拓扑的G蛋白偶联受体跨膜螺旋三维结构的预测方法与流程
本发明涉及G蛋白偶联受体结构预测技术领域,尤其涉及一种基于结构拓扑的G蛋白偶联受体跨膜螺旋三维结构的预测方法。
背景技术:
2012年两位美国科学家因在G蛋白偶联受体(G-protein-coupledreceptors,下文简称GPCR)的卓越研究获得了诺贝尔化学奖。研究GPCR的主要意义在于:它是人体内最大的信号传导蛋白质超家族,可以与激素、神经递质、光、气味分子等小分子物质发生相互作用,在细胞信号传递中发挥着重要作用。人类重大疾病的发生往往都与GPCR功能紊乱有关。据统计,目前世界前200个最畅销药物中超过10%都是以GPCR为靶点。GPCR是嵌在生物膜上的一类特殊的膜蛋白,一般需要与生物膜结合才能形成稳定的天然(Native)构象,这使得用核磁共振(NMR,NuclearMagneticResonance)和X射线晶体衍射(X-ray)方法来获得GPCRs的三维结构变得异常困难。这也导致三维结构的GPCRs数量远落后于序列测定的数量。目前,人类基因组大约已经测定了800个左右的GPCR序列,解构的目标只有近10个。虽然用计算机方法预测GPCR的三维结构是最理想的方法,也取得了很大的进展,但是由于GPCR极少的先验知识,使得这仍是一项极具挑战性的研究。由于GPCR结构的特殊性且稀缺解构构象,通用的球蛋白结构预测方法很难直接应用于GPCR的三维结构预测。GPCR三维结构预测方法通常可以分为三类:同源建模(homology-basedmodeling,也称比较建模)、穿线法(threading)、从头预测(denovomodeling)。同源建模方法的核心思想是通过目标序列的同源蛋白质来推定其三维结构,其关键步骤是将序列相似的模板片段,直接复制给候选结构,这样的方法虽然简单可行,但忽视了螺旋结构的多样性。穿线法的核心思想是寻找和目标序列没有显著性同源关系、但是具有同一折叠(fold)类型的蛋白质,并建立模块库。因此,用穿线法生成的跨膜螺旋区的精度主要受模板库中螺旋模板精度的影响。Zhang等人(参见文献ZhangY,DeVriesME,andSkolnickJ.Structuremodelingofallidentifiedgprotein-coupledreceptorsinthehumangenome.PLoSComputBiol,2006,2(2):88–99)是穿线法的倡导者与领军人物。从头预测法是从氨基酸序列出发,通过GPCR的先验知识与规则,对目标结构的采样不依赖于模板,在所有可能的空间中全范围离散采样,用能量函数对所有可能的结果逐一评价,这样的采样方法无疑是耗时巨大的。Baker等人(参见文献Yarov-YarovoyV,SchonbrunJ,BakerD.Multipassmembraneproteinstructurepredictionusingrosetta.Proteins,2006,62:1010–1025与文献BarthP,SchonbrunJ,BakerD.Towardhigh-resolutionpredictionanddesignoftransmembranehelicalproteinstructures.ProcNatlAcadSciUSA,2007,104:15682–15687)与Goddard等人(参见文献GoddardWA,KimSK,LiY,etal.Predicted3Dstructuresforadenosinereceptorsboundtoligands:Comparisontothecrystalstructure.JournalofStructuralBiology,2010,170:10–20)在使用从头预测方法时,都遇到了类似的困难。上述这些方法本质是遇到了同一个问题:难以在七个跨膜螺旋结构的保守性与局部多样性之间获得平衡,其实质是未将两者统一到一个系统模型中。
技术实现要素:
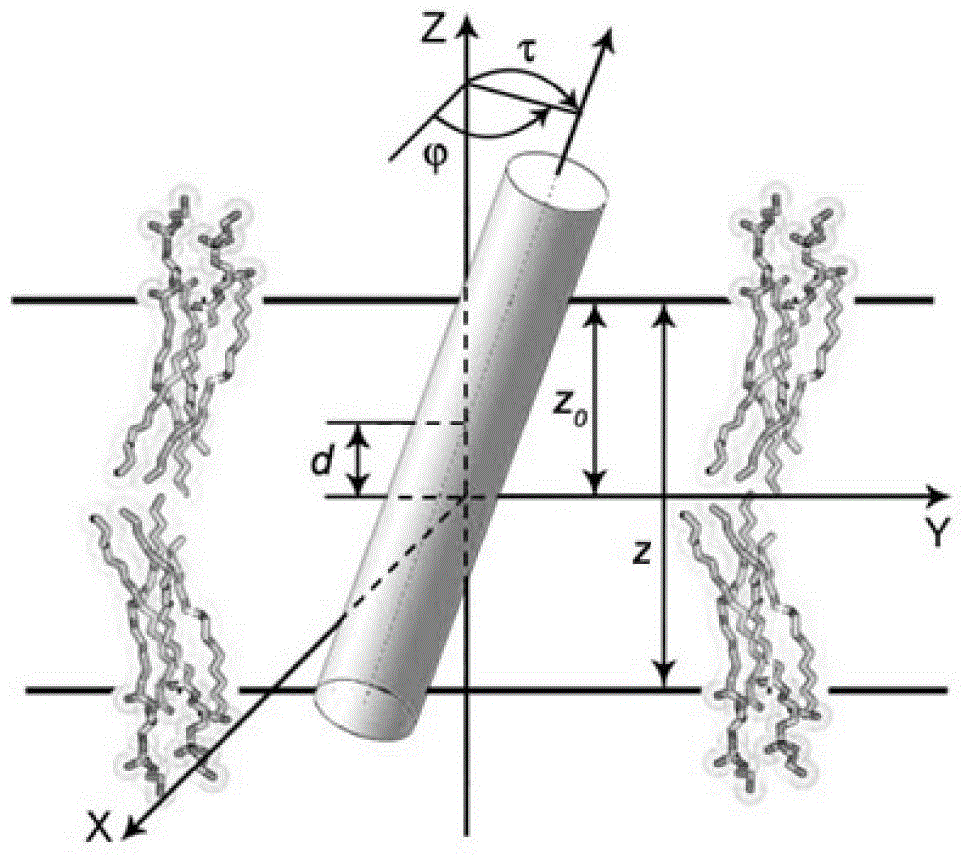
本发明所要解决的技术问题是提供一种基于结构拓扑的G蛋白偶联受体跨膜螺旋三维结构的预测方法,本发明方法在现有的并行多模板预测方法patGPCR(参见文献WuH,LüQ,QuanL,etal.PatGPCR:AmultitemplateApproachforImproving3DStructurePredictionofTransmembraneHelicesofG-protein-coupledReceptors,ComputationalandMathematicalMethodsinMedicine,inpress)的框架下,跨膜螺旋的三维结构预测为目标,建立基于结构的拓扑模型,并利用该模型形成了四阶段的结构优化方法,同时引入基于拓扑结构的能量项与约束,起到了优化评判标准与剪裁采样空间的作用。本发明方法中的跨膜螺旋三维结构预测方法称为TMGPCR(structuralpredictionforTransMembranehelixesofGPCR)。为解决以上技术问题,本发明采用如下技术方案:一种基于结构拓扑的G蛋白偶联受体跨膜螺旋三维结构的预测方法,所述预测方法包括:(1)构建跨膜螺旋的结构拓扑模型:先构建G蛋白偶联受体的7个跨膜螺旋的几何结构拓扑模型,所述跨膜螺旋的几何结构拓扑模型包括螺旋与螺旋之间的结构拓扑、螺旋与膜之间的结构拓扑;(2)预测所述跨膜螺旋的结构拓扑模型,分为以下四个阶段:(21)第一阶段为识别跨膜螺旋区域:根据G蛋白偶联受体的序列信息预测出可能的跨膜螺旋区域;(22)第二阶段为优化跨膜螺旋间的相对位置:通过对第j个螺旋在空间位置上的平移,利用能量函数e对G蛋白偶联受体构象进行能量计算,选择能量最小的G蛋白偶联受体构象,其中,1≤j≤7;(23)第三阶段为优化每个螺旋在膜内的自转朝向:通过对所述第j个螺旋以螺旋轴为中心轴自转i度,利用能量函数e对G蛋白偶联受体构象进行能量计算,选择能量最小的G蛋白偶联受体构象;(24)第四阶段为优化每个螺旋与膜的倾斜角度:对膜平面法线与所述第j个螺旋的螺旋轴的夹角进行优化,利用能量函数e对G蛋白偶联受体构象进行能量计算,选择能量最小的G蛋白偶联受体构象;(3)Loop重建:用Loop片段插入连结所述7个螺旋,最后将G蛋白偶联受体整体结构进行小幅度的优化。优选地,步骤(1)中所述螺旋与螺旋之间的结构拓扑是所述螺旋在所述膜平面上的垂直投影,体现所述螺旋与螺旋之间的2D距离关系。优选地,步骤(1)中所述螺旋与膜之间的结构拓扑体现所述螺旋与所述膜平面之间的几何关系。优选地,步骤(21)中识别跨膜螺旋区域的方法为:综合六种主流跨膜螺旋识别方法,将所述六种主流跨螺旋识别方法的结果的平均值作为所述识别跨膜螺旋区域,所述六种主流跨螺旋识别方法为TopPred、UniProt、TMpred、HMMTOP、TMHMM、OCTOPUS。优选地,对步骤(22)中优化跨膜螺旋间的相对位置引入2D位置约束,即所述7个螺旋的中心线与所述膜平面的交点位置约束,以文献NugentT,JonesdDT.Predictingtransmembranehelixpackingarrangementsusingresiduecontactsandaforce-directedalgorithm.PLoSComputBiol,2009,6:e1000714的预测方法预测出的结果作为所述2D位置约束。优选地,对步骤(24)优化每个螺旋与膜的倾斜角度引入倾斜角度约束,即所述螺旋相对所述膜平面法线的倾斜角度,以满足30度为均值、6度为方差的高斯分布作为在所述倾斜角度优化时的约束。优选地,所述能量函数e为用于评价跨膜螺旋之间位置合理性的能量函数E与用于评价螺旋堆积成螺旋束时的紧密程度的能量函数E′之和,即e=E+E′,所述能量函数E为Rosetta的膜环境能量函数,所述能量函数E′为关于跨膜螺旋间的能量函数,当所述螺旋之间不发生碰撞时,E′越小,螺旋束越紧密,形成的构象就越合理;当所述螺旋之间发生碰撞时,所述能量函数E远远大于所述能量函数E′,所述能量函数E′相对于所述能量函数E来说可以忽略不计,所述能量函数e由能量函数E进行评价。具体地,所述能量函数E′由式(1)计算得到,Smax---(1)]]>式(1)中,S为所述螺旋与螺旋之间的结构拓扑模型中的任意两个螺旋中点与这些中点的均值坐标所构成三角形的面积的面积和,Smin为从现有已知G蛋白偶联受体中得到的面积最小值,Smax为从现有已知G蛋白偶联受体中得到的面积最大值。由于采用以上技术方案,本发明与现有技术相比具有如下优点:本发明的预测方法针对跨膜螺旋的空间结构特点,建立了兼顾保守性与多样性的结构拓扑模型,并利用该模型形成了四阶段的结构优化方法,试图获得采样广度与深度的平衡。同时,引入基于结构拓扑的能量项与约束,起到了优化评判标准和剪裁采样空间的作用,有效的预测了跨膜螺旋的三维结构。本发明通过与三组经典数据集的比较实验检验了本发明方法预测GPCR跨膜螺旋三维结构的有效性。附图说明图1为螺旋与螺旋之间的结构拓扑图;图2为螺旋与膜之间的结构拓扑;图3为GPCR建模流程图;图4为本发明方法中使用的2D位置约束(CXCR4);图5为本发明方法中使用的2D位置约束(D3);图6为TMGPCR与Swiss的TMRMSD与TotalRMSD比较;图7为TMGPCR的50折叠bootstrap与Swiss比较(TMRMSD比较);图8为TMGPCR的50折叠bootstrap与Swiss比较(TotalRMSD比较)。具体实施方式下面结合附图来进一步阐述本发明。一种基于结构拓扑的G蛋白偶联受体跨膜螺旋三维结构的预测方法,包括:(1)构建跨膜螺旋的结构拓扑模型本发明方法所指的的结构拓扑(StructuralTopology)有别于计算机图论中的拓扑结构(Topology),结构拓扑在本发明方法中指GPCR三维结构的拓扑,在此简称为结构拓扑。大多数的GPCR都具有类似的结构拓扑:七个α跨膜螺旋(TMH,TransMembraneHelix)、一个N端、三个胞内环(ICL,IntraCellularLoop)、三个胞外环(ECL,ExtraCellularLoop)以及一个C端。其中,七个α跨膜螺旋所构成的螺旋束是其最主要的拓扑特征,决定了GPCR的整体走向,其三维结构的预测精度直接影响受体三维结构预测、配体对接及功能分析的准确性。对于受体结构预测的后继问题-配体对接(LigandDocking),配体口袋(Pocket)柔性与对接柔性的模拟都与螺旋束的结构有关。用计算机方法对GPCR的跨膜螺旋进行预测时,我们首先建立7个跨膜螺旋的几何结构拓扑模型,建模的重点是跨膜螺旋之间的几何结构拓扑特征,以及跨膜螺旋与膜平面之间的几何关系。此时,不考虑单个螺旋内部原子之间的相对位置,将单个螺旋视为刚体(RigidBody)。结构拓扑模型是对跨膜螺旋实际三维结构的简化与归纳,通过拓扑模型的分析,可以设计出有效的结构采样策略,并协助评估不同采样策略的性能。本发明方法中跨膜螺旋的结构拓扑有两类:螺旋与螺旋之间的结构拓扑(如图1所示)和螺旋与膜之间结构拓扑(如图2所示)。如图1为螺旋在膜平面上的垂直投影,体现了螺旋与螺旋之间的2D距离关系。如图2模型中Z表示膜的厚度,Z=2z0,其中,表示螺旋的旋转角度,τ表示螺旋的相对膜平面法线的倾斜角度,d是螺旋中心相对于膜平面的垂直偏移。(2)基于结构拓扑模型的预测方法(21)预测方法流程依据对结构拓扑模型的分析,本发明的跨膜螺旋的预测方法可以分为四个阶段。第一阶段:跨膜螺旋区域识别,根据GPCR的序列信息预测出可能的跨膜螺旋区,这是进行跨膜螺旋三维结构预测的必要先导步骤;第二阶段:从7个螺旋2D起点位置开始,建立螺旋的3D结构,并通过平移优化螺旋间的相对位置关系;第三阶段:优化各个螺旋在膜内的自转朝向,即找到最佳的旋转角度第四阶段:优化7个螺旋与膜的倾斜角度,即找到最佳的倾斜角度τ。第二、三、四阶段先后对螺旋执行平移、自转、倾斜三种运动,这是依据GPCR的结构拓扑而设计的,缺少了任意一种运动,螺旋采样都是不完整的。所采用的分阶段优化方法,不仅是采样代价与采样搜索空间的平衡,更是横向搜索与纵向搜索的一种平衡,为了保持GPCR结构预测的完整性,螺旋结构预测完成后,用Loop片段插入连结7个螺旋,最后将整体结构进行小幅度的优化,其流程如图3所示。(22)基于结构拓扑的打分函数能量函数是蛋白质三维结构预测中的重要子问题,优质的能量函数能较准确的识别近天然构象,指导采样算法向正确的方向搜索;拙劣的能量函数即使采样到高精度构象,也会失之交臂。本发明方法在Rosetta的能量函数基础上,设计了基于拓扑结构的能量项,用于评判跨膜螺旋之间位置合理性的能量项。Rosetta是著名的从头预测平台,在历届CASP(CriticalAssessmentofTechniquesforProteinStructurePrediction)比赛中都名列前茅,他们设计了针对膜环境的能量函数(即打分函数,蛋白质结构预测领域常称为能量函数),将厚度的膜分为7层,统计了28个膜蛋白中各种残基在7层中的分布概率,用此能量函数E来评估膜蛋白的三维结构。这样的能量函数考虑了螺旋残基与膜之间的相对位置,但未对跨膜螺旋之间位置关系进行评价。本发明方法在Rosetta的膜环境能量函数E上增加了一个新的、关于跨膜螺旋的能量项E′用于评价螺旋堆积成螺旋束时的紧密程度,即本发明方法中的用于计算G蛋白偶联受体构象能量的能量函数为e,且e=E+E′。一般认为,在不发生碰撞前提下,越是紧密的螺旋束就越接近天然构象。E′先计算图1中任意两个螺旋中点与这些中点的均值坐标O所构成三角形的面积S1,S2,…,S7的面积和S,然后通过公式(1)计算E′。其中Smin与Smax是从现有已知GPCR中得到的面积最小值与最大值。在不发生碰撞情况下,E′越小,螺旋束越紧密,形成的构象就越合理;在发生碰撞时,能量函数E远远大于能量函数E′,能量函数E′相对于能量函数E来说可以忽略不计,能量函数e由Rosetta的膜环境能量函数E进行评价。实际情况中各螺旋的中点并不在同一平面上,且可能发生三角形相交的情况,但由于公式(1)中计算的为相对量,所以这并不影响构象评价的可靠性,式(1)如下所示,Smax---(1).]]>(23)引入约束依据上述跨膜螺旋的拓扑结构模型,将模型中的各种参数进行离散化,然后按排列组合采样,若没有其它限制条件,无疑其采样空间巨大,是一个典型的NP困难问题。由此在预测的各个阶段,需要引入多种约束,剪裁采样空间,从而寻求计算代价与预测质量之间的平衡。约束数据可以来源于生化实验、第三方预测结果或是先验知识。本发明方法引入了两种约束。第一种是2D位置约束,即7个螺旋的中心线与膜平面的交点位置约束。这种约束限制了7个螺旋在2D平面上的相对关系,可以避免远离这些约束点的无效采样。本发明方法以Nugent等人(参见文献NugentT,JonesdDT.Predictingtransmembranehelixpackingarrangementsusingresiduecontactsandaforce-directedalgorithm.PLoSComputBiol,2009,6:e1000714)的预测方法预测出的结果作为2D位置约束,该方法首先预测出脂在膜中的暴露程度、残基与残基和螺旋与螺旋之间的相互作用,然后用Force-Directed算法优化膜蛋白中跨膜螺旋的二维拓扑结构。以GPCRDOCK2010比赛目标CXCR4和D3为例,用Nugent等人的方法计算得到的2D位置约束,如图3-4所示,具体螺旋坐标值如表1所示。表1中helixnumber、CXCR4x、CXCR4Y、D3x、D3Y分别表示螺旋序号、CXCR4的x轴坐标、CXCR4的Y轴坐标、D3的x轴坐标、D3的Y轴坐标。第二种是倾斜角度约束,本发明从OMP(OrientationsofProteinsinMembranesdatabase)数据库中获取了倾斜角度τ的先验知识,OMP数据库中存放了1980个己知膜蛋白的倾斜角度等三维拓扑信息,具有统计意义。GPCR是膜蛋白家族中的一种具有7个跨膜螺旋的膜蛋白,所以OMP中的数据对GPCR具有一定的适用性。经对OMP中数据统计,发现螺旋的倾斜角度τ满足以30度为均值,6度为方差的高斯分布约束,由此TMGPCR以此作为在螺旋倾斜角度优化时的约束。表1CXCR4和D3的螺旋中心2D坐标helixnumberCXCR4XCXCR4YD3XD3Y117.015-4.15114.2303.57228.3277.431-2.02612.5603-7.0412.165-8.690-0.5384-6.02011.7207.15910.9705-10.846-6.659-14.79011.98663.758-9.971-5.122-14.13072.8110.4220.4960.045(24)识别跨膜螺旋区域跨膜区域的识别虽然也是跨膜螺旋中一个必要步骤,但现有方法相对较成熟,所以本发明综合了现有的六种主流跨膜螺旋识别方法,将这些识别结果的平均值作为本发明的识别区域。表2是以目标CXCR4为例,用TopPred、UniProt、TMpred、HMMTOP、TMHMM、OCTOPUS进行预测的结果,以及本发明平均后的结果和天然构象的实际跨膜螺旋区域。表2CXCR4的跨膜区域识别方法7个跨膜区域(起始残基号-终止残基号)TopPred9-29,43-63,77-97,120-140,169-189,205-225,252-272UniProt4-28,43-64,76-95,120-139,161-181,207-226,248-267TMpred4-27,43-70,79-98,120-140,169-188,205-226,251-270HMMTOP4-26,45-64,79-97,120-142,169-188,207-226,253-272TMHMM4-26,47-69,79-101,121-140,164-186,207-229,249-271OCTOPUS6-26,41-61,71-101,118-138,166-186,205-225,247-267Result6-27,43-65,76-101,119-139,166-186,206-226,250-269Native2-27,41-65,71-97,119-139,161-186,206-230,248-266(25)基于结构拓扑模型的采样与算法在本发明的第二、三、四阶段中,将7个螺旋作为独立刚体,对这些刚体之间的相对位置进行优化。刚体是内部原子相对位置保持不变,只能进行整体移动或旋转的结构,这样的移动符合单个跨膜螺旋的内部残基较为稳定,自由度较低的特点。以下通过算法描述第二、三、四阶段的具体过程。其中,第3行是迭代优化螺旋的结构,每次迭代以前一代的最优构象为初始构象;第4行getHelixAxis()函数以7个螺旋中的骨架原子坐标为输入,分别计算它们的中心轴的方向向量;第5行getMembrane()函数将7个螺旋轴方向的均值作为膜平面的法线方向;第二阶段为第6-11行,其中transferHelix(P,TM[j],RND(-5,5),RND(-5,5),RND(-5,5))函数对第j个螺旋分别沿x、y、z方向随机(在至范围取随机整数)平移,若平移后的构象能量函数e打分低于原构象的打分,则接受该构象;第三阶段为第12-17行,其中spinHelix(P,TM[j],i)函数表示对第j个螺旋以螺旋轴为中心轴自转i度,并用能量函数e评估是否接受自转后的构象;第四阶段为第18-23行,其中tiltGaussianHelix(P,TM[j],30,5)函数对膜平面法线与第j个螺旋轴的夹角τ进行均值为30度和方差为6度的高斯采样,并用能量函数e评估是否接受采样后的构象;第24行crossHelixes()函数执行7个螺旋的两两交换,并从中选出最优构象。以下通过与三组经典数据集的比较实验,检验本发明方法预测GPCR跨膜螺旋三维结构的有效性。实验一与GPCRDOCK2010参赛结果比较我们考察了TMGPCR对GPCRDOCK2010参赛小组所提交构象的优化能力。实验中先将网上公布的提交结构按照TMRMSD排序,从中分别按照TMRMSD均匀选取了结构,其中CXCR4的最小TMRMSD为D3的最小TMRMSD为用本发明方法优化后所得到的最小TMRMSD分别为和在GPCRDOCK2010网站公布的结果中分别排名为第四和第一,如表3中所示GPCRDOCK2010中的前五名的构象与TMGPCR产生的最优构象的TMRMSD比较。表3与GPCRDOCK2010参赛结果的TMRMSD比较实验二与Swiss方法的双盲比较为了检验TMGPCR预测GPCR跨膜螺旋结构的性能,进行了一组双盲实验,即在仅已知GPCR序列情况下进行预测。双盲方式也是GPCRDock2008/2010、CASP(TheCriticalAssessmentofproteinStructurePrediction)等计算机结构预测比赛中采用的方法。实验目标是GPCRDock网站上公布的所有解构目标,共八个。同时用这八个目标分别在著名的同源建模服务网站Swiss上进行预测。TMGPCR与Swiss使用相同的模板,每个目标选取2-4个模板,如表4所示,Swiss预测中有三个模板没有输出结果。TMGPCR每个目标平均产生1358.75个构象,产生每个目标平均0.32个CPU小时。TMGPCR与Swiss的结果用盒须图进行比较,按TMRMSD从小到大排序后取TMGPCR的前400个,如图6所示,图中灰色与黑色分别表示TMGPCR的TMRMSD与整体RMSD(TotalRMSD)的最小值、1/4位数值、3/4位数值和最大值,标记+与×分别表示TMGPCR结果集的中位数值与平均值,标记○表示Swiss的结果。图6中TMGPCR获得的最小TMRMSD低于Swiss的目标有五个,分别是CXCR4、KOR1、D3、A2A和Beta2;两个目标的最小TMRMSD与Swiss结果相当,分别是HH1R和S1P1;KOR3的最小TMRMSD明显劣于Swiss的结果。从图6中还发现有四个目标的最小TotalRMSD低于Swiss的结果KOR1、HH1R、KOR3和Beta2;两个目标的最小TotalRMSD与Swiss的结果相当,分别是A2A与S1P1;CXCR4和D3的最小TotalRMSD劣于Swiss的结果。这说明TMGPCR不仅提高了跨膜螺旋部分的精度,而且还利用螺旋位置的调整改善了Loop的结构精度,从而提高了目标的整体预测精度。随后,考察了TMGPCR结果集的统计学意义。使用bioshellpackage(如参见文献GrontD,KolinskiA.Bioshell-apackageoftoolsforstructuralbiologycomputations.Bioinformatics,2006,22:621–622)对TMGPCR结果集的前400个结构进行1%的50折叠(fold)bootstrap估计,然后将bootstrap后的均值分别与Swiss的21个结果进行TMRMSD与TotalRMSD的比较,如图7所示,X轴表示Swiss的TMRMSD的值,Y轴表示TMGPCRbootstrap后的TMRMSD均值,所以图右下方的点表示TMGPCR优于Swiss,有16个,用+表示;左上方表示TMGPCR劣于Swiss,有2个,用×表示;斜线附近(均值差<0.05)则认为两者质量相当,有3个,用⊙表示。每个点的上下沿表示bootstrap后方差,方差越小TMGPCR结果的质量越稳定,反之则质量越不稳定。由于图7与图8的刻度比例为5:11,所以将图8中的方差放大11/5倍,以使得两张图的方差宽度具有相同意义。同样的方法比较了TMGPCR与Swiss的TotalRMSD情况,TMGPCR也具有优势,如图8所示。实验三与MODELLER方法的双盲比较MODELLER也是一个被广泛使用的同源建模工具,而且同时支持单模板(single-template,ST)与多模板(multiple-template,MT)建模。故本实验在相同的硬件与软件平台上开展了TMGPCR与MODELLER的性能比较实验。实验中,单模板与多模板的MODELLER分别对表4中的每个GPCR目标产生1300个候选构象,它们结果集的TMRMSD均值与方差显示在表4的第8和第9列中。从均值来看,与单模板MODELLER(第8列)的比较TMGPCR(第6列)在八个目标中有六个取得了优势;与多模板MODELLER(第9列)的比较中,TMGPCR有七个目标取得了优势。从方差来看,多模板MODELLER普遍方差较低,这一方面说明多模板MODELLER的稳定性较高;另一方面也说明多模板MODELLER采样时的多样性不足,仅在模板的较近的空间采样,这也可能是导致多模板MODELLER预测精度偏高的原因。表4八个目标选取的模板与候选集精度比较上述表4中,(a)TMGPCR产生的候选集的构象数量。(b)TMGPCR产生的候选集构象的平均TMRMSD与方差。(c)ST(single-template,单模板)MODELLER产生的候选集构象的平均TMRMSD与方差。(d)MT(multiple-template,多模板)MODELLER产生的候选集构象的平均TMRMSD与方差。以上对本发明做了详尽的描述,其目的在于让熟悉此领域技术的人士能够了解本发明的内容并加以实施,并不能以此限制本发明的保护范围,且本发明不限于上述的实施例,凡根据本发明的精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 5‑HT2B拮抗剂的制造方法与工艺
- 1‑(噻唑‑2‑基)哌啶‑4‑醇的合成方法与制造工艺
- 一种大麻酚类化合物的合成方法与制造工艺
- 一类对GPR84具有激动作用的化合物及其制备方法和用途与制造工艺
- eGFP标记GPR120阳性细胞的小鼠模型构建方法
- 靶向生长抑素受体的Al<sup>18</sup>F?NOTA?PEG<sub>6</sub>?TATE及其制备方法和应用
- 吡咯烷衍生物的口服制剂的制作方法
- 胰高血糖素样肽-1受体激动剂制备治疗肺动脉高压药物的应用
- 一种armc5基因检测试剂盒及突变位点的制作方法
- 作为mglu5受体的负性别构调节剂的6-氟-2-[4-(吡啶-2-基)丁-3-炔-1-基]咪唑并[1,2a ...的制作方法