一种环状RGD肽及其虚拟筛选方法与流程
本发明涉及一种环状RGD肽及其虚拟筛选方法。
背景技术:
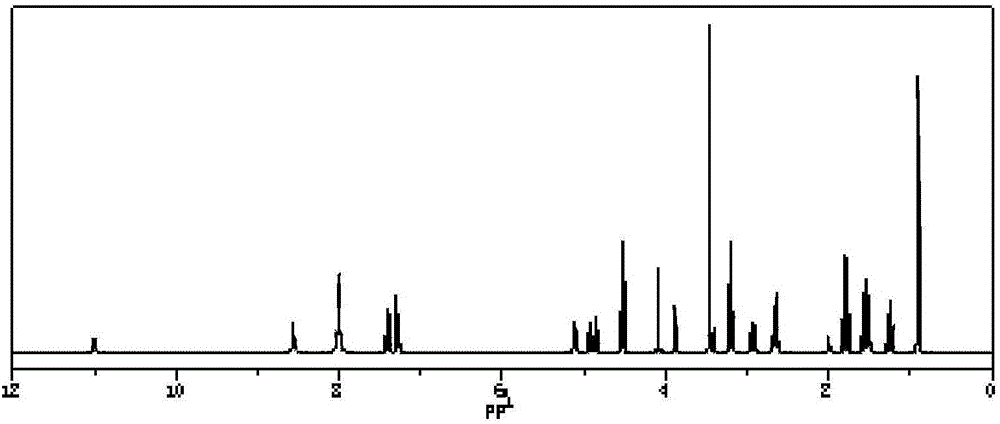
:血管生成是指在已有血管的基础上生成新血管,血管生成是身体的正常发育,繁殖和组织修复的基础,也是许多慢性和潜在的致命疾病的发病机制。超过70种的疾病都与血管生成有关,包括肿瘤的生长和转移、糖尿病性视网膜病、脉络膜新生血管和类风湿性关节炎等。然而在临床上,治疗上述疾病尚存在很多缺点:例如,化疗是癌症治疗的最常见的方法;然而,在一般情况下,化疗药物具有低的选择性和显著的副作用,这限制了癌症治疗的效果。又如玻璃体内注射是治疗脉络膜新生血管形成的最常见的方式,然而这种方式患者的接受程度低,且需要多次重复的玻璃体内注射,还可增加眼损伤的几率。因此,迫切需要开发新的和有效的治疗方法,加强新生血管性疾病治疗和增强患者的依从性。整合素αvβ3,是由α亚基和β亚基经非共价键连接而形成的异二聚体跨膜糖蛋白粘附分子,在许多重要的病理生理过程中起到重要的作用,如细胞增殖、分化、侵袭和迁移、凋亡、组织修复和肿瘤的侵润转移等。在成熟血管内皮细胞和绝大多数正常器官系统不表达或表达极少,而在肿瘤、炎症等组织改建时表达明显增加。整合素αvβ3的异常高表达,可以通过与多种其它细胞因子间的协同作用,促进肿瘤血管形成,并且促进肿瘤的侵袭及转移。因此,特异性的干扰或破坏整合素αvβ3分子的功能,可以抑制表达αvβ3分子的新生血管内皮细胞的凋亡,使细胞因缺乏营养而死亡,从而达到抗肿瘤细胞生长及转移的作用,被视为抗肿瘤治疗和血管新生相关疾病的靶点之一。RGD肽是一类含有精氨酸-甘氨酸-天冬氨酸(Arg-Gly-Asp)序列的短肽,1984年Pierschbacher首次报道了纤维蛋白原中含有的RGD序列为细胞识别位点。之后的研究发现,外源性RGD多肽及其类似物可与体内含RGD序列的物质竞争结合,从而阻断血管上皮细胞增殖的信使传递,终止细胞增殖,使血管不能生长,导致肿瘤组织供氧系统中断,最终细胞萎缩、凋亡。基于上述原因,大量药物开发工作集中于RGD和相关肽,已有RGD的药物在临床试验中。RGD序列肽可选择性的靶向于整合素αvβ3,起到整合素受体拮抗的作用。多数线形RGD肽在体内循环的半衰期较短,进一步的现有技术中公开了环形RGD肽类,其更加稳定,且较线形RGD多肽有更高的受体结合特异性。现有技术公开了氨基酸序列RGDfK、RGDfV等环状RGD序列肽,但其与整合素αvβ3受体亲和力不高。因此,有必要提出有效的技术方案,解决上述问题。技术实现要素:本发明针对上述现有技术的不足,提供一种。本发明提供的环状RGD多肽分子可以克服上述血管生成疾病治疗中的缺点,可选择性的靶向于整合素αvβ3,起到整合素受体拮抗的作用,又适合于将药物、基因治疗载体或其他试剂选择性靶向至合适组织。本发明解决上述技术问题的技术方案如下:一种环状RGD肽的虚拟筛选方法,步骤如下:(1)虚拟筛选过程:以RGD三肽为先导化合物,通过计算机辅助药物设计软件Surflex-dock,采用Hammerhead打分函数打分,从合成的现实角度出发,综合静电作用、范德华力、疏水作用和氢键进行打分,根据打分结果排序,初步确定环状RGD肽化合物;(2)对接过程:从蛋白质数据库PDB中下载编号1L5G蛋白,编号1L5G蛋白包括RGD肽和αvβ3,去除其中的RGD肽,将1L5G蛋白中剩余的αvβ3与步骤(1)中确定的环状RGD肽进行对接,筛选出打分高的六元环肽。本发明还提供一种上述环状RGD肽,所述环状RGD肽具有RGDfVK、RGDfKV、RGDKfV或RGDf(N-me)VK氨基酸序列。优选的,所述环状RGD肽为式I所示的c(RGDf(N-me)VK),或式II所示的c(RGDf(N-me)VK)-C。优选的,所述环状RGD肽为式III所示的c(RGDfVK),式IV所示的c(RGDfVK)-C,或式V所示的c(RGDfVK)-GDK。优选的,所述环状RGD肽为式VI所示的c(RGDfKV),式VII所示的c(RGDfKV)-C,或式VIII所示的c(RGDfKV)-GD。优选的,所述环状RGD肽为式IX所示的c(RGDKfV),或式X所示的c(RGDKfV)-C。有益效果:本发明环状RGD肽,可以与整合素αvβ3受体的特异性结合作用,可以作为血管生成拮抗剂,也适用于将药物、基因治疗载体或其他试剂选择性靶向至合适组织,作为低毒、高效的疾病治疗和诊断的方式。通过本发明虚拟筛选方法得到的环状RGD肽,与已知环肽相比具有较好的活性,与整合素受体亲和力强。附图说明后文将参照附图以示例性而非限制性的方式详细描述本发明的一些具体实施例。附图中相同的附图标记标示了相同或类似的部件或部分。本领域技术人员应该理解,这些附图未必是按比例绘制的。附图中:图1为本发明c(RGDf(N-me)VK)的模拟核磁图谱;图2为本发明c(RGDf(N-me)VK)-C的模拟核磁图谱;图3为本发明c(RGDf(N-me)VK)-C的液相图谱;图4为本发明c(RGDf(N-me)VK)-C的质谱;图5为本发明c(RGDfVK)的模拟核磁图谱;图6为本发明c(RGDfVK)-C的模拟核磁图谱;图7为本发明c(RGDfVK)-GDK的模拟核磁图谱;图8为本发明c(RGDfKV)的模拟核磁图谱;图9为本发明c(RGDfKV)-C的模拟核磁图谱;图10为本发明c(RGDfKV)-GD的模拟核磁图谱;图11为本发明c(RGDKfV)的模拟核磁图谱;图12为本发明c(RGDKfV)-C的模拟核磁图谱;图13为本发明应用例1中RGD肽与PLGA-PEG-Mal的连接1HNMR图谱;图14为本发明应用例1中RGD肽与PLGA-PEG-Mal的连接IR图谱;图15为本发明应用例1中姜黄素纳米粒透射电镜图;图16为本发明应用例1中细胞摄取的定量考察图;图17为本发明应用例1中细胞摄取的定性考察图;图18为本发明应用例1中荧光显微镜观察细胞摄取图;图19为本发明应用例1中细胞的生长抑制图;图20为本发明应用例1中不同时间点荷瘤裸鼠体内荧光分布图;图21为本发明应用例1中离体组织器官荧光强度成像图;图22为本发明荧光显微镜下观察细胞摄取情况图。具体实施方式针对传统药物或治疗方式的缺点,本发明开发了一种环状RGD肽,具有与整合素αvβ3受体的特异性结合作用,可以作为血管生成拮抗剂,也适用于将药物、基因治疗载体或其他试剂选择性靶向至合适组织,作为低毒、高效的疾病治疗和诊断的方式。本发明一种环状RGD肽的虚拟筛选方法,步骤如下:虚拟筛选过程:以RGD三肽为先导化合物,通过计算机辅助药物设计软件Surflex-dock,采用Hammerhead打分函数打分,从合成的现实角度出发,综合静电作用、范德华力、疏水作用和氢键进行打分,根据打分结果排序,初步确定环状RGD肽化合物;对接过程:从蛋白质数据库PDB中下载编号1L5G蛋白,编号1L5G蛋白包括RGD肽和αvβ3,去除其中的RGD肽,将1L5G蛋白中剩余的αvβ3与步骤(1)中确定的环状RGD肽进行对接,筛选出打分最高的具有RGDfVK、RGDfKV、RGDKfV或RGDf(N-me)VK氨基酸序列的六元环肽,具体为本发明式I至式10所示的c(RGDf(N-me)VK)、c(RGDf(N-me)VK)-C、c(RGDfVK)、c(RGDfVK)-C、c(RGDfVK)-GDK、c(RGDfKV)、c(RGDfKV)-C、c(RGDfKV)-GD、c(RGDKfV)、c(RGDKfV)-C,对接结果打分列表如表1所示:表1本发明环肽对接打分表环肽对接打分c(RGDf(N-me)VK)7.98c(RGDf(N-me)VK)-C10.81c(RGDfVK)7.46c(RGDfVK)-C8.97c(RGDfVK)-GDK7.54c(RGDfKV)8.72c(RGDfKV)-C9.35c(RGDfKV)-GD7.69c(RGDKfV)7.36c(RGDKfV)-C7.78已知文献报道的活性环肽c(RGDyK)的打分为7.11,如表1所示,本发明环肽与已知环肽相比具有较好的活性。图1、图2、图5至图12所示,为本发明六元环肽的模拟核磁图谱。本发明环状RGD肽可以通过标准的化学反应合成。例如,一种肽或多肽可以被合成并且不从其合成树脂上切割下,而一种肽或蛋白的另一片段可以被合成并且随后从所述树脂上切割下,从而暴露出在所述另一片段上被功能阻断的末端基团。通过肽缩合反应,此两个片段可经分别在它们的羧基端和氨基端的肽键共价地连接,以形成其片段。将含有本发明所述序列的氨基酸片段直接收尾相接环化形成环状RGD肽。通过赖氨酸作为衔接,可以在赖氨酸端通过酰胺键继续引入氨基酸或氨基酸片段。如通过酰胺键在环状RGD肽的赖氨酸上接入半胱氨酸,由于半胱氨酸上含有活泼巯基,该巯基可进一步通过化学反应与载体材料、药物和荧光染料缀合,形成缀合物或组合物。以下多肽合成方法以环状c(RGDf(N-me)VK)-C肽为例,该方法同样适用于本专利所述的其他环状RGD序列肽的合成(改变氨基酸及其连接顺序)。缩写:Arg(R),精氨酸;Cys(C),半胱氨酸;DCM,二氯甲烷;Dde,1-(4,4-二甲基-2,6-二氧代亚环己基)乙基;DIEA,二异丙基乙胺;DMF,N,N-二甲基甲酰胺;Fmoc,芴甲氧羰基;Gly(G),甘氨酸;HBTU,O-苯并三唑N,N,N,N-四甲基-六氟磷酸;Lys,赖氨酸;Pbf,2,2,4,6,7-五甲基-二氢苯并呋喃-5-磺酰基;RP-HPLC,反相高效液相色谱;RT,室温;TFA,三氟乙酸;Val(V),缬氨酸。(1)、溶胀树脂:将2-氯三苯甲基氯树脂放入反应管中,加二氯甲烷,振荡30min;(2)、天冬氨酸-树脂连接:通过沙芯漏斗抽滤掉溶剂,加入3倍摩尔过量的Fmoc-Asp(OAll)-OH,再加入10倍摩尔过量的二异丙基乙胺,最后加入N,N-二甲基甲酰胺溶解,振荡30min;甲醇封头,30min;(3)、脱保护:去掉溶剂,加20%哌啶/DMF溶液(15ml/g),5min;去掉再加20%哌啶/DMF溶液(15ml/g),15min;抽掉哌啶溶液,取树脂,加入茚三酮,氰化钾,苯酚溶液各一滴,检测反应进行性;(4)、偶联:洗涤后,取保护氨基酸Fmoc-Gly-OH三倍过量,O-苯并三唑N,N,N,N-四甲基-六氟磷酸三倍过量,均用少量DMF溶解,加入反应管,加入二异丙基乙胺十倍过量,反应30min;(5)、清洗后重复上述步骤依次连接Fmoc-Arg(Pbf)-OH,Fmoc-phe-OH,Fmoc-N-Me-Val-OH,Fmoc-Lys(Dde)-OH,脱除最后一个氨基酸的Fmoc保护基;(6)、去除Dde保护基:2%水合肼/DMF(15ml/g)两次各15min;洗树脂,缩合氨基酸Fmoc-Cys(Trt)-OH,去除Fmoc保护基;(7)、切割、纯化,冻干得环状c(RGDf(N-me)VK)-C肽。如图3和图4所示,对产品做MS的分子量鉴定,和HPLC分析的纯度鉴定。液相检测条件:Columan:4.6*150mm,kromasilC18-5;SolventA:0.1%Trifluoroaceticin100%Acetonirile;SolventB:0.1%Trifluoroaceticin100%Water;Gradient:0.01minA5%,B95%;25.0minA70%,B30%;Flowrate:1.0ml/min;Wavelength:214nm;Volume:10ul.质谱条件:Flowrate:0.2ml/min;RunTime:1min;BufferA:0.1%HCOOHinwater;BufferB:0.1%HCOOHinAcetonirile.(1)流式细胞仪评价本发明环状RGD肽与整合素受体亲和力荧光细胞率的检测:取整合素高表达的对数生长期HUVEC细胞,以每孔1×105个细胞接种于6孔板,培养24h后,弃去培养基,每孔加入2mL含10ug·mL-1姜黄素浓度的纳米粒培养基,继续培养;同时不加药的空白孔作为对照。30min后取出6孔板,消化,转移至1.5mL的EP管中。1000r·min-1离心5min,弃上清,PBS洗3次,再加入600μL的PBS吹打均匀,进样,流式细胞仪检测荧光细胞率。表2本发明环肽荧光细胞率表环肽荧光细胞率(%)c(RGDf(N-me)VK)7.98c(RGDf(N-me)VK)-C10.81c(RGDfVK)7.46c(RGDfVK)-C8.97c(RGDfVK)-GDK7.54c(RGDfKV)8.72c(RGDfKV)-C9.35c(RGDfKV)-GD7.69c(RGDKfV)7.36c(RGDKfV)-C7.78表2为流式细胞仪检测荧光细胞率,由流式结果可知,本发明环肽与整合素受体具有较好的亲和力。(2)选取得分高的本发明3个环状RGD肽(c(RGDf(N-me)VK)-C,4c(RGDfVK)-C,c(RGDfKV)-C)开展对人脐静脉细胞摄取的研究。取对数生长期的HUVEC细胞接种于24孔板,每孔中加一片圆形盖玻片,每孔加入1ml培养基,每孔接种密度为细胞数40000个,细胞培养箱培养24h至细胞贴壁。吸弃旧的培养基,每孔加入1ml含20ug·ml-1姜黄素浓度的不同环肽的纳米粒培养基,继续培养,于细胞培养箱中孵育2h。弃去培养基用PBS洗3次,加入4%的多聚甲醛溶液室温固定15min,再用PBS清洗3次,取出盖玻片倒扣于载玻片上,缓冲甘油封片,于荧光显微镜下观察细胞摄取情况。由图22的荧光图片可见,所制备的环肽纳米粒的荧光强度强于对照环肽,与对照环肽相比具有更高的亲和力。应用例1六元环肽作为靶向受体或荧光探针在肿瘤治疗和诊断的应用研究将六元环肽与高分子载体通过静电吸附或共价键相结合,可携带化疗药物,将药物选择性的靶向输送到肿瘤组织局部,可降低化疗药物的毒副作用。(1)RGD肽高分子载体缀合物的制备本发明所述RGD肽高分子缀合物包括并不限于所述的载体材料。RGD肽与PLGA-PEG-Mal的连接(选择实验例中得到的较高亲和力的肽c(RGDf(N-me)VK)-C):称取PLGA-PEG-Mal,丙酮溶解,氮气吹干使其均匀分散,加入PBS(pH7.4)过夜;称取与PLGA-PEG-MAL等摩尔的RGD肽,纯化水溶解,滴加到PLGA-PEG-Mal溶液中,充氮,冰浴,搅拌,HPLC监测反应进程;反应完成后,超滤离心清洗除去杂质,冷冻干燥即得RGD-PEG-PLGA。RGD-SH与PLGA-PEG-Mal在pH7.4~7.6的PBS条件下发生迈克尔加成反应,生成RGD-PEG-PLGA。HPLC色谱检测结果显示,PLGA-PEG-Mal链接RGD肽的效率约为90%。图13为其1HNMR图谱,其中B中δ=6.705(2H)为-Mal-的特征峰,链接上RGD肽以后,其-Mal-特征峰消失;A中δ=1.22,δ=2.08和δ=4.16为RGD肽的特征峰,与PLGA-PEG-Mal的特征峰重合。PLGA-PEG-Mal、RGD肽和RGD-PEG-PLGA的IR表征如图14所示,图14中的A:在δ=3516.77cm-1附近为OH吸收峰,δ=2925.54cm-1、2855.04cm-1为甲基和亚甲基C-H伸缩振动吸收峰,δ=1759.97cm-1为C=O的伸缩振动吸收峰,δ=1454.91cm-1为C-C的一系列伸缩振动吸收峰,δ=1170.63cm-1为酯键的C-O伸缩振动吸收峰,δ=1095.12cm-1为PEG中醚键C-O-C的伸缩振动吸收峰。B:δ=3333.39cm-1为羧基中OH的伸缩振动吸收峰,δ=1662.59cm-1为酰胺键的C=O伸缩振动吸收峰。C:连接RGD后,RGD中羧基OH的吸收峰由于周围化学环境的改变,使其发生移动至δ=2961.69cm-1处;其它的峰由于分子量比较小,与PLGA-PEG-Mal的峰重合。(2)姜黄素纳米粒的制备及表征称取合成高分子和姜黄素,丙酮溶解为有机相,将有机相滴加到水相,搅拌挥发除去有机溶剂,即得姜黄素纳米粒。激光粒度仪测定纳米粒的粒径及粒度分布;如图15所示,透射电子显微镜观察纳米粒的形态。(3)细胞水平的靶向性评价如图16和图17所示,采用流式细胞仪进行细胞摄取的定量考察(浓度和时间依赖性);如图18所示,荧光显微镜进行细胞摄取的定性考察(时间依赖性)。由结果可见,靶向组的纳米粒荧光强度强于普通纳米粒组。(4)姜黄素纳米粒对细胞的抑制作用考察待细胞在培养皿中长成紧密的单层后,用0.25%胰蛋白酶消化,用含10%胎牛血清的DMEM培养基配成单个细胞悬液,分别以每孔3000个细胞接种于96孔培养板中,每孔体积100ul。将培养板放入37℃、5%CO2的细胞培养箱中培养24h,弃去旧的培养基,每孔加入100ul不同姜黄素浓度(1.25、2.5、5、10、20、30、40ug·ml-1,对应的材料浓度为30、60、120、240、480、720、960ug·ml-1)的培养基,每个浓度4个平行孔,同时不加药的空白孔作为对照,接着放入恒温培养箱中继续培养48h。然后每孔加入20ulMTT溶液(5mg·ml-1),继续孵育4h,终止培养。吸弃孔内培养液,每孔加入200ulDMSO,摇床振荡10min,使甲瓒充分溶解。选择570nm波长,在酶联免疫检测仪上测定各孔光吸收值,记录结果。计算细胞存活率:细胞存活率(%)=实验组OD值/对照组OD值×100%。如图19所示,细胞的生长抑制图。由实验结果可知,靶向纳米粒的细胞存活率最低,其对细胞的生长抑制作用强于普通纳米粒和药物溶液。(5)动物水平的靶向性评价A549细胞以每只5×106个接种于裸鼠右前腿上方,肿瘤长至800mm3左右时用于实验。取雄性荷瘤裸鼠6只,分成3组,每组2只。分别尾静脉注射0.2mL载有荧光素DiR的RGD-PEG-PLGA/DiR-Np,mPEG-PLGA/DiR-Np和荧光素DiR的甲醇水溶液,分别于1,2,4,8,12和24h水合氯醛麻醉,记录小鼠的荧光成像照片。并于荧光最强的点取出心、肝、脾、肺、肾及肿瘤,进行离体组织器官成像,使用KodakMIinvivoFxPro处理图像和数据分析。如图20所示,不同时间点荷瘤裸鼠体内荧光分布图。如图21所示,离体组织器官荧光强度成像图.给药后不同时间,荷瘤裸鼠体内荧光分布情况如图20所示,药物通过尾静脉注射入体内后首先在肝脏聚集,这可能与肝脏网状内皮系统巨噬细胞的吞噬有关。RGD-PEG-PLGA/DiR-Np组在2h肿瘤处有微弱的荧光,8h荧光强度达到最大,随后逐渐降低;mPEG-PLGA/DiR-Np组在各个时间点的荧光强度都弱于靶向组;DiR甲醇水溶液组主要在肝脏聚集。给药后8h取出心、肝、脾、肺、肾及肿瘤,可见RGD-PEG-PLGA/DiR-Np组在肿瘤组织的荧光强度明显强于mPEG-PLGA/DiR-Np组,DiR甲醇水溶液组只有微弱的荧光。靶向纳米粒组在肝、肾等器官的荧光强度弱于普通纳米粒组。通过这种方法不仅可以实现肿瘤的靶向治疗,还可以接上荧光探针作为肿瘤诊断使用。应用例2六元环肽作为靶向受体或荧光探针在眼后段疾病治疗中的应用通过建立BN大鼠的CNV模型、考察PAMAM-PEG-RGD(TAT)地塞米松纳米复合物与PAMAM-PEG地塞米松纳米复合物在CNV模型BN大鼠和健康BN大鼠的药代和组织分布。1、CNV动物模型的建立动物:棕色雄性BN大鼠(北京维通利华实验动物中心,体重180-220g)。仪器:氩激光机(LUMENIS公司,型号VISONONE),荧光眼底血管造影(FFA)摄像机(TOPCON公司,日本,型号TRC-50IX)试剂:荧光素钠注射液(广西梧州制药股份有限公司、3ml:0.6g,血管造影)、复方托品酰胺滴眼液(长春迪瑞制药有限公司、5ml/支,散瞳),1%甲基纤维素滴眼液。麻醉(10%水合氯醛,4ml/kg麻醉状态良好);双眼滴用复方托品酰胺滴眼液散瞳,实验眼滴用1%甲基纤维素(防止进一步刺激的保护剂);左眼前放置90度的前置镜,应用532nm氩激光仪经裂隙灯显微镜在全视网膜镜下围绕视盘等距光凝几个点,光斑直径75μm,激光功率70mW,曝光时间0.1s,以光凝后光凝斑中央有气泡形成或轻度出血为bruch膜破裂的标志。FFA检查:于光凝后3、7、14、21、28及56天分别随机抽取挪威大鼠麻醉并散瞳,腹腔注射20%的荧光素钠注射液,实验眼和对照眼分别行FFA检查(FFA的圆盘状荧光渗漏可证实CNV存在,21天有荧光渗漏的光凝斑数达高峰)。2、药品地塞米松磷酸钠注射液(济南利民制药有限责任公司,产品批号14100625-1);PEG-PAMAM地塞米松纳米复合物;PEG-PAMAM-RGD(TAT)地塞米松纳米复合物。3、药代和组织分布取已建模的BN大鼠和健康BN大鼠各42只,分别分为A、B、C三组:A:地塞米松磷酸钠注射液B:PEG-PAMAM地塞米松纳米复合物C:PEG-PAMAM-RGD(TAT)地塞米松纳米复合物分好组的大鼠,每组14只,称重。分别尾静脉注射PEG-PAMAM地塞米松纳米复合物、PEG-PAMAM-RGD(TAT)地塞米松纳米复合物和地塞米松磷酸钠注射液(10.0mg/kg),给药后于5min、30min、1、2、4、12、24h的每个时间点眼眶取血后断颈处死大鼠2只,迅速解剖取眼、心、肝、脾、肺、肾等组织脏器,将每个时间点每组2只小鼠的血和脏器混合,各脏器分别称重,血液按照小鼠体重8%计算。取将各脏器匀浆和血浆各0.3ml置于离心管中,加入乙腈200μl,涡旋3min后,12000r/min离心10min,取上清液HPLC分析。计算每个时间点地塞米松的血药浓度和组织分布。色谱条件色谱柱:DiscoveryC-18(4.6×250mm,5μm)流动相:甲醇:水=70:30(v/v)检测波长:240nm流速:1mL/min柱温:30℃进样量:20μL地塞米松在生物样品中的浓度与其被检测到的峰面积具有较好的线性关系,符合生物样品分析的要求。大鼠体内药时数据用药代动力学程序进行处理。4、实验结果:药物在大鼠组织和血浆中的药物浓度见表3-表8。表3:健康BN大鼠地塞米松磷酸钠注射液在血浆和组织中的药物浓度(μg/g、μg/ml)表4:CNV模型大鼠地塞米松磷酸钠注射液在血浆和组织中的药物浓度(μg/g、μg/ml)时间(h)眼心肝脾肺肾血0.085.16.140.35.810.314.98.60.58.97.528.96.112.111.217.9110.29.76.64.26.24.526.222.96.919.83.15.911.916.341,53.41.62.24.24.511.9120.79.40.50.51.95.05.224nd1.711.1ndndndnd表5:健康BN大鼠PAMAM-PEG地塞米松纳米复合物在血浆和组织中的药物浓度(μg/g、μg/ml)时间(h)眼心肝脾肺肾血0.083.35.975.225.55.43.93.30.53.96.350.116.02.27.16.813.212.428.923.48.13.839.622.09.720.114.13.310.430.340.85.116.36.91.915.127.712nd8.06.05.40.72.76.024nd2.29.92.8ndnd0.5表6:CNV模型大鼠PAMAM-PEG地塞米松纳米复合物在血浆和组织中的药物浓度(μg/g、μg/ml)时间(h)眼心肝脾肺肾血0.086.26.757.317.33.85.34.10.511.45.462.520.16.43.83.216.714.441.921.14.92.333.923.08.720.117.52.34.241.142.73.114.42.70.82.130.3121.4nd7.82.2nd0.77.424ndnd10.40.5ndnd1.0表7:健康BN大鼠PAMAM-PEG-RGD(TAT)地塞米松纳米复合物在血浆和组织中的药物浓度(μg/g、μg/ml)时间(h)眼心肝脾肺肾血0.083.15.769.036.66.63.14.20.53.75.847.320.93.36.37.212.88.834.530.15.85.233.421.410.227.718.14.87.740.540.95.813.58.02.210.730.312nd8.23.72.30.51.95.124nd1.81.4ndndnd0.9表8:CNV模型大鼠PAMAM-PEG-iRGD(TAT)地塞米松纳米复合物在血浆和组织中的药物浓度(μg/g、μg/ml)时间(h)眼心肝脾肺肾血0.087.94.520.113.13.44.92.40.520.25.631.810.94.22.72.9143.95.922.48.34.83.527.4232.23.912.49.83.32.244.6421.31.46.15.12.01.127.91214.3nd2.71.7ndnd13.9245.6nd0.8ndndnd4.2表9:药代参数由上述实验结果可见:1)地塞米松磷酸钠注射液BN大鼠和CNV造模的BN大鼠尾静脉注射后,药物迅速在大鼠各器官和血液中分布,在健康大鼠和CNV大鼠中主要分布在心、肝、肾中,眼中分布较少,但相比健康大鼠由于CNV的形成眼部脉络膜有泄漏和血管增生导致药物在眼部的积聚有所增加。2)PAMAM-PEG地塞米松纳米复合物BN大鼠和CNV造模的BN大鼠尾静脉注射后,药物迅速在大鼠各器官和血液中分布,在健康大鼠主要分布在肝、脾、肾中,眼中分布较少;在CNV大鼠中主要分布在肝、脾、心中,在眼中分布也较少,但相比健康大鼠由于纳米粒具有一定的被动靶向作用,在有EPR效应的眼部眼部的积聚有所增加,但由于血脑屏障的存在,这种被动靶向的作用并不明显。3)PAMAM-PEG-RGD(TAT)地塞米松纳米复合物BN大鼠和CNV造模的BN大鼠尾静脉注射后,药物迅速在大鼠各器官和血液中分布,在健康大鼠主要分布在肝、脾、肾中,眼中分布较少;在CNV大鼠中主要分布在眼、肝、脾中,说明RGD和TAT共同介导的PAMAM聚合物纳米复合物可以透过血脑屏障并靶向到整合素受体高表达的疾病部位,可用于该疾病的靶向治疗。4)制成纳米复合物后,地塞米松在血浆中的半衰期明显延长,血中生物利用度明显增加。但靶向纳米复合物与普通纳米粒相比生物利用度没有明显差别,但在眼组织分布上明显增加,同时由于眼部药物的增多,药物在其他脏器的积聚降低,这也降低了药物对正常器官的毒副作用。本发明获得的环状RGD肽具有更高的受体结合性能和靶向选择性。可直接注射使用作为新生血管拮抗剂治疗肿瘤、脉络膜新生血管等疾病。也可作为靶向载体,携带药物、基因、荧光标记物等特异性的与患病组织或细胞结合,起到疾病治疗和诊断的作用。本发明获得的环状RGD肽可应用于抗血管生成剂、癌症化学治疗剂、细胞毒剂、抗炎剂、纳米颗粒、多肽、核酸分子、小分子、荧光团、荧光素、罗丹明、放射性核素或它们的结合物。其中抗血管生成剂包括但不限于甲磺酸阿帕替尼(ApatinibMesyIate)、重组人血管内皮抑制素、贝伐单抗(Bevacizumab)、pegaptanib、雷珠单抗(ranibizumab)、索拉非尼(Sorafenib)。癌症化学治疗剂包括但不限于:5-氟脲嘧啶、甲氨蝶呤、阿糖胞苷和环胞苷、长春碱、长春新碱、喜树碱、羟基喜树碱、紫杉醇、多烯紫杉醇、阿霉素、依托泊甙、拓扑替康、吉西他滨、顺铂、卡铂。抗炎剂包括但不限于:氢化可的松、可的松、强的松、地塞米松、倍他米松、曲安奈德、乙酸阿奈可他、姜黄素、葛根素、穿心莲内酯或汉防己甲素。本发明可用其他的不违背本发明的精神或主要特征的具体形式来概述。因此,无论从哪一点来看,本发明的上述实施方案都只能认为是对本发明的说明而不能限制本发明,权利要求书指出了本发明的范围,而上述的说明并未指出本发明的范围,因此,在与本发明的权利要求书相当的含义和范围内的任何改变,都应认为是包括在本发明的权利要求书的范围内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1