一种基因序列比对的云计算加速方法与流程
本发明涉及生物基因数据处理领域,尤其是指一种基因序列比对的云计算加速方法,具体是基于云计算框架加速通用基因序列比对程序的方法。
背景技术:
随着基因二代测序技术(nextgenerationsequencing,ngs)的发展,单个基因测序成本已下降至1000美元以下。与此同时,基因测序的数据正呈现爆炸式增长,以illuminahiseqxtmten为例,一次运行可以产生60亿条序列信息。有关数据表明,每6个月基因数据量就会翻一番,而依照这个增长速度,到2020年,每年产生的基因数据将达到1个exabase(每4个base等于1个字节),而2025年,这个数据将增长至每年1个zettabase。基因测序数据量的增长及成本的降低都在以一个远超摩尔定律的速度在发展,如何快速处理所产生的基因数据正面临严峻的挑战。
无论采用何种测序技术,基因序列比对分析都是理解测序结果数据的最重要一步,也是目前耗时最长步骤之一。序列比对是将测序所得的短序列与已有的参考基因序列进行序列比对,寻找测序结果在参考基因序列上的精确定位。在二代测序技术中,根据测序方法的不同,又可分为单端测序(single-read)与双末端测序(paired-end/mate-paired)两类。单端测序时将基因组随机打断,再对每个片段进行测序,最终只产生单个测序结果文件。双端测序则是对一个长的序列测得其两端序列,最终将产生互成一对的两个测序结果文件。现在的大多数二代测序平台都采用fastq格式保存下机数据,测序时的一条读序(read),在fastq文件中将以序列名,序列,“+”,质量分数四行数据描述。在双端测序得到的两个fastq文件中,序列名相同,位置上一一对应的两条读序巧好为测序时同一个基因片段的两端序列。常用于对基因二代测序数据进行比对的工具有bowtie2、bwa、maq、soap2等等,其中又以bwa软件在基因序列比对中应用的最为广泛。而这些成熟的基因序列的比对工具都有两个共同特点,即仅可在单台计算机上运行。当面对二代测序技术的高速发展,人们更希望能在多机上运行序列比对程序,以获取更高的并行效率及扩展性,使基因序列数据的处理可以在一个合理的时间范围内完成。目前在这个问题上已有的解决方案可大致分为以下两种:
一是采用mpi方式在多机上运行序列比对程序,例如pbwa和pmap。而如人们所知,mpi的效率虽高,但开发难度极大,且任何一个节点出问题都将导致整个任务的失败,容错上更多的需要人为保证。同时mpi的性能优化更多是针对超算硬件进行优化,而对于云上所采用的常用硬件,mpi做的优化通常不会有太大效果。
二是在spark框架上采用jni调用bwa软件,如sparkbwa及gatk4.0工具包中的bwaspark都是采用了这类方案。相比于mpi解决方案,spark方法的容错性由spark框架本身保证,开发难度较低,但jni的方式开发难度不小,且性能一般,有实验显示java调用jni空函数与java调用java空方法性能存在5倍左右的差距,而额外的性能优化还需要开发者对jvm机制有足够的了解。
本发明将采用一种更简单的方式将比对工具运行在spark框架上,不仅可以很好的利用spark的机制进行多机计算的调度,数据的分发、监控和容错,而且相较于jni的实现方式,开发门槛低,代码维护简单,性能更好,扩展性可接近线性。同时,本发明的结构设计具有松耦合的特点,无需修改代码,就可灵活更换基因序列比对程序。除此之外,本发明对数据预处理步骤进行了优化,大大降低了数据预处理的的耗时。
技术实现要素:
本发明的目的在于克服现有技术的不足,提供了一种基因序列比对的云计算加速方法,该方法基于大数据技术,在多节点上运行序列比对程序的框架,以改善当前基因序列数据比对耗时较长的问题,为后续基因数据分析、压缩、组装打下坚实基础。
为实现上述目的,本发明所提供的技术方案为:一种基因序列比对的云计算加速方法,包括以下步骤:
1)对基因测序仪的下机数据文件fastq进行预处理,以保证数据分发时数据的完整性;
2)通过spark对修改后的基因测序数据完成多节点分发;
3)对每个节点所获得的修改后的基因数据,恢复其原有fastq文件格式;
4)每个节点通过spark中的pipe算子执行基因序列比对程序脚本,运行结果存储在spark的弹性分布式数据集rdd(resilientdistributeddatasets)中;
5)运行结果保存在诸如hdfs、amazon、s3等分布式文件系统。
在步骤1)中,对基因测序仪的下机数据文件fastq进行预处理包括读取数据,修改与合并多个输入文件及将数据保存至分布式文件系统或共享文件系统;
所述修改与合并多个输入文件,包括以下步骤:
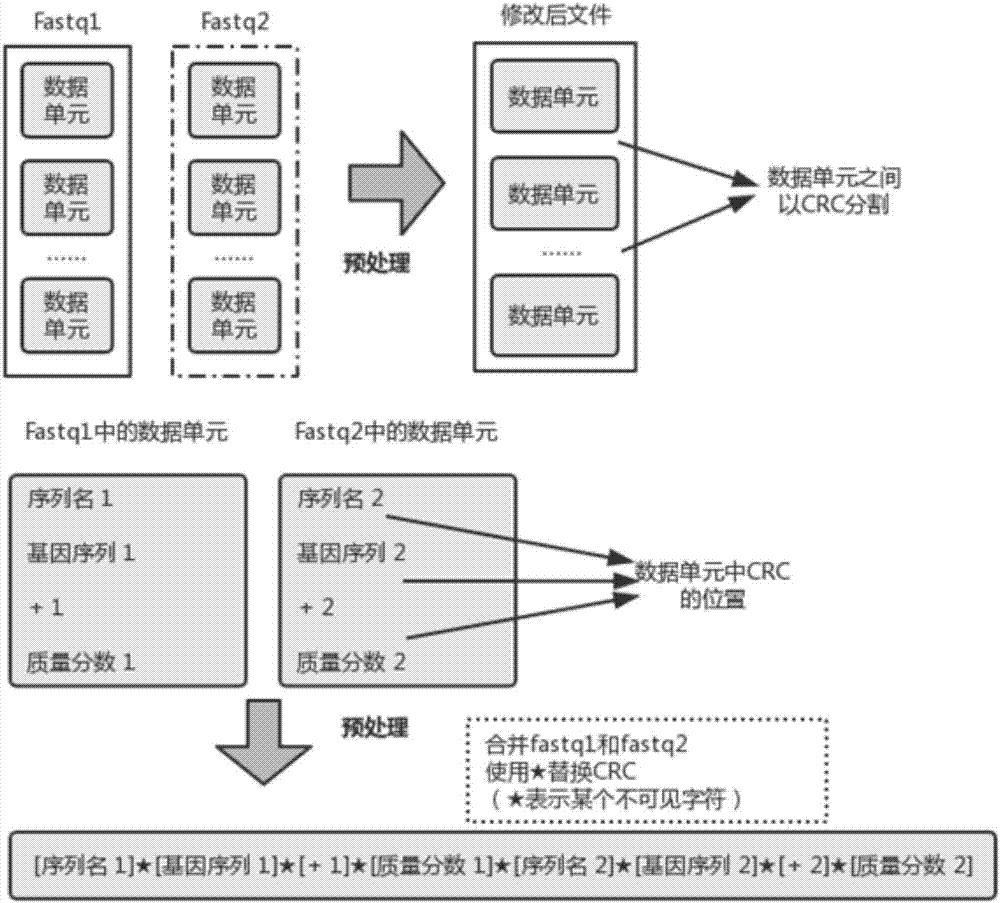
1.1)对于fastq文件,在一个数据单元内,统一使用不可见字符替换所有的回车字符,不同数据单元之间,以回车字符进行标记,其中,将以fastq文件中每四行组成的一条读序完整信息称作fastq文件的一个数据单元,而对于双末端测序,成对的读序称为修改后文件的一个数据单元;
1.2)将两个fastq文件中一一对应的数据单元相互连接;
1.3)重复步骤1.1)、1.2),直至fastq文件处理完毕,并生成一个新的数据文件;
在步骤2)中,针对步骤1.1)中以回车字符为不同数据单元间的标记这前提,选用按行读取的sparkapi对数据进行读取,sparkapi将会承担对数据进行多节点分发的操作。
所述读取数据,合并多个输入文件及将数据保存至分布式文件系统hdfs的预处理步骤采用流水线模式优化。
在步骤3)中,对每个节点的处理数据恢复原有数据形态,以下操作二选一:
①将所有数据中的不可见字符重新恢复为回车字符,恢复后的中间数据写入各个节点本地硬盘;
②将所有数据中的不可见字符重新恢复为回车字符,恢复后的中间数据保存在spark的rdd中。
在步骤4)中,所述pipe算子实现在spark中调用外部程序处理rdd中数据,处理后数据保存在新的rdd中,其中,pipe算子调用的外部程序即为基因序列比对程序,其参数为基因序列比对程序执行脚本的保存路径。
所述基因序列比对程序为bwa。
在步骤5)中,使用hadoopapi将含有基因序列比对程序运行结果的rdd保存于分布式文件系统。
本发明与现有技术相比,具有如下优点与有益效果:
本发明方法使用了spark中的pipe算子将基因序列比对程序运行在spark环境中,并同时实现了将中间数据写入硬盘和中间数据存储在rdd中的两种可选方式,相较于只可运行于单节点的基因序列比对程序,如bwa,本发明可以处理更大量的数据;相对于mpi等实现方式,本发明的调度,容错机制更易开发与维护;相对于现有使用jni方式实现的基于spark框架加速bwa的方法,本发明实现更为简单,代码更易维护;相对于现有的其他多节点实现,本发明的性能更高,扩展性、兼容性、灵活性更好,更容易集成新的单节点比对程序,进而可以从下层的改进中获益。
同时,本发明方法还对数据预处理步骤采用了流水线模式进行优化,相对于现有的多采用串行方式对基因序列比对数据进行预处理的方式,能大大缩减了预处理步骤的耗时。
附图说明
图1为本发明的使用spark框架加速基因序列比对方法流程图。
图2为本发明的合并多个文件的方法流程图。
图3为本发明的预处理步骤采用流水线优化方法流程图。
具体实施方式
下面结合具体实施例对本发明作进一步说明。
如图1所示,本实施例所提供的基因序列比对的云计算加速方法,包括以下步骤:
s1、对基因测序仪的下机数据文件fastq进行预处理,以保证数据分发时数据的完整性,包括读取数据,合并多个输入文件及数据保存至文件系统。
图2中给出了fastq格式文件的样式及修改后的文件形式。fastq文件中以每四行组成一条读序的完整信息,即图2中的fastq文件内的一个数据单元。双端测序会产生两个fastq文件,即图2中的fastq1和fastq2两个文件。两个fastq文件中的数据单元一一对应,共同组成基因序列比对程序所需读入的一个完整信息,即图2中的修改后文件的一个数据单元。需要保证在稍后的spark自动数据分发过程中,每个spark分区(partition)得到的数据都以一个完整信息为单位。
所述步骤s1包括以下几个分步骤:
s1.1、读取数据:从本地文件系统中读取双端测序后的fastq1和fastq2两个文件。
s1.2、修改与合并多个输入文件,过程如下:
s1.2.1、对于fastq文件,在一个数据单元内,统一使用某个的不可见字符替换所有的回车字符,不同数据单元之间,以回车字符进行标记。
先从fastq1文件中取出四行数据,并将四行数据中的所有回车字符更换为ascii码值为6的不可见字符,再对fastq2文件重复在fastq1文件中的操作。处理好的数据单元在文件中以回车字符进行数据单元间的标记。
s1.2.2、将两个fastq文件中一一对应的数据单元相互连接。
对于两个fastq文件中一一对应的数据单元,两者间的回车字符也替换为ascii码值为6的不可见字符,由此生成修改后文件的一个数据单元。
s1.2.3、重复s1.2.1与s1.2.2步骤,直至两个fastq文件处理完毕,并生成一个新的数据文件。
s1.3、数据保存至文件系统:使用hdfs的putapi,将新生成数据上传至hdfs。
进一步地,以上读取数据、修改与合并多个输入文件、数据保存至文件系统三个步骤可采用流水线模式处理。如图3所示,将上述三个部分放入不同的进程中去执行,以减少运行时间。
1)readfile进程:用于读取fastq文件,并对单个文件进行字符替换,生成fastq文件中的数据单元。
使用readfile进程先读取fastq文件的一部分,即图3中的数据1(数据块的大小设置应结合网络带宽考虑),并完成对数据1的修改,修改后数据传给merge进程,readfile进程继续读取fastq文件下一个数据块2;
2)merge进程:对两个fastq文件中的数据单元进行合并,生成修改后文件数据单元;
merge进程接收到readfile进程传过来的数据1后,对两个文件的对应数据单元进行合并,形成合并文件块,并将新生成的数据块传给upload进程,随后,merge进程继续处理readfile进程新传入的数据块2。
3)upload进程:将合并后的数据上传至分布式文件系统hdfs。
upload进程接收到merge进程传过来的数据1后,将其写到分布式文件系统hdfs上,然后继续等待接收merge进程下一次传过来的数据2,再将数据2使用hdfs提供的appendapi,追加至文件的末尾,完成后继续等待merge进程传递新的数据块。
以上预处理步骤可使用java,python等语言实现,只要hdfs提供了相应接口。
s2、对数据进行多节点分发
通过spark的textfile()方法读入已存储在hdfs上修改后的文本文件,创建rdd1。textfile()方法有两个参数值,一个是修改后文本文件的路径名,一个是spark分区大小设置,可以对分区大小采用默认值,即与hdfs的一个块(block)的大小相等,spark会将数据按照已设定的分区大小情况,自动均衡分布至多个节点。
s3、对每个节点所获得的数据,恢复其原有形态,以下操作二选一:
①将所有数据中的不可见字符重新恢复为回车字符,恢复后的中间数据写入各个节点本地硬盘。
单独撰写一个脚本a,实现从标准输入流中读取数据,对读入数据中所有ascii码值为6的字符替换为回车字符,并将新数据每8行中的前四行数据存入fastq1,后四行数据存入fastq2中,即恢复原有的双端测序中两个fastq文件的格式。文件命名可采用时间+进程号+_1.fastq及时间+进程号+_1.fastq形式,保存在各个节点本地硬盘上,再以标准输出流形式输出其路径名。
spark程序中,对rdd1使用pipe算子(一个可在spark程序中调用外部程序处理rdd中数据的算子),调用脚本a,pipe的参数即为脚本a路径名。此时恢复后的数据文件存储在了各个节点的本地硬盘上。新生成的rdd2中保存着各个数据文件的路径名。
②将所有数据中的不可见字符重新恢复为回车字符,恢复后的中间数据保存在rdd中。
对rdd1使用spark的flatmap算子,以ascii码值为6的字符为数据分割标记,生成rdd2,此时rdd2中保存的即为恢复后的数据。
方法①可最大限度保证对各类基因比对程序的兼容性,方法②可用于支持从标准输入流读入数据的基因比对程序(如bwa),实现简单,此方法性能略好于①。
s4、每个节点通过pipe算子执行基因序列比对程序执行脚本
将运行基因序列比对程序bwa所需的shell脚本(bwa运行脚本的书写参考bwa软件使用手册)路径作为pipe参数。新生成的rdd3中保存了bwa软件的运行结果。
s5、运行结果保存在每个节点本地分布式文件系统hdfs
使用spark的saveastextfile()方法,将rdd3中数据保存在hdfs上。
以上所述实施例只为本发明之较佳实施例,并非以此限制本发明的实施范围,故凡依本发明之形状、原理所作的变化,均应涵盖在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!