一种ctDNA低频突变测序数据分析方法和装置与流程
本发明属于测序技术领域,具体而言,涉及一种ctDNA低频突变测序数据分析方法和装置。
背景技术:
ctDNA(circulating tumorDNA),又名循环肿瘤DNA,是肿瘤细胞释放到血液循环系统中的DNA,可以准确反映原发肿瘤组织的分子遗传学信息,通过检测ctDNA可以获得点突变、结构变异,甚至染色体拷贝数变异等遗传信息。基于肿瘤的异质性,血液里ctDNA含量极少,癌症早期ctDNA丰度甚至达到0.1%以下的水平,这导致基因突变频率通常介于0.01%~1%之间(属于低频突变)。
新一代测序技术(NGS)是目前应用最广的测序技术,具有测序深度高、通量大、准确率高、灵敏度好等优势,然而NGS应用于ctDNA低频突变检测仍存在技术难点。一方面,NGS不可避免的存在测序误差,单碱基错误率一般在0.1%~1%之间,另一方面,测序文库构建一般使用高保真酶进行PCR扩增,也存在10-6左右的复制错误率,并且随着PCR循环数增多而增大;这两方面的因素导致ctDNA低频突变测序分析时存在较大的背景噪音,特别是在0.1%及以下检测限的情况下,将难以区分模板DNA突变与测序错误/复制错误,从而容易出现假阳性的分析结果。
为解决上述问题,通常采用两种做法:一是提高测序数据量,通过加大数据量来提高测序深度,从而排除测序错误,然而数据量的增加和测序深度并非线性相关,纠错效果非常有限;二是分子标签法,通过在原始DNA分子的至少一端连接分子标签,分子标签可以是一段随机碱基组成的核苷酸序列,长度按照实际需要选择,基于分子标签长度和碱基的变化,理论上,分子标签可以有4n种,并且经过分子标签标记的原始DNA分子是独一无二的;但实际上,分子标签合成及标记DNA分子时依然存在偏好性问题,不可避免会出现优势分子标签,并且通常会出现多个DNA分子标记同一个分子标签的情况,这导致ctDNA低频突变检测过程中,增加了识别原始DNA分子的难度,并且检测准确率难以保证,因此,仍需要对基于分子标签的ctDNA低频突变测序分析方法进行改进,以提高检测准确率,避免假阳性结果的出现。
技术实现要素:
本发明的目的在于提供一种ctDNA低频突变测序数据分析方法和装置。
本发明所采取的技术方案是:
一种低频突变测序数据分析方法,包括:
S1:获取基于分子标签构建的扩增子文库测序下机数据,所述下机数据包括读段和分子标签;
S2:根据读段与参考基因组的比对结果进行质控;
S3:将具有相同读段的分子标签集合为第一标签群;
S4:在第一标签群内,根据频数和分子标签之间的编辑距离,找出聚类的类别中心;
S5:在第一标签群内,将非类别中心的分子标签归类于相似性最高的类别中心,形成具有相似标签序列的第二标签群;
S6:在第二标签群内,构建一致性序列,根据一致性序列与参考基因组的比对结果进行突变分析,输出突变结果。
作为上述方法优选的,步骤S4进一步包括:
S41:在第一标签群内,统计分子标签的频数;
S42:针对频数为L的当前分子标签,找出与当前分子标签编辑距离≤阈值范围的分子标签集合;如果集合内没有序列,则当前分子标签作为一个类别中心;否则,将当前分子标签的频数均分到集合内频数≥5L的分子标签的频数上,当前分子标签的频数则归0;
S43:对步骤S42进行重复递归,遍历每条分子标签;
S44:将频数>0的分子标签作为类别中心。
作为上述方法优选的,步骤S5进一步包括:
S51:在第一标签群内,找出与非类别中心的分子标签编辑距离最小的类别中心,将所述分子标签归类于所述类别中心,形成具有相似标签序列的第二标签群;
S52:如果同一分子标签归类于两个及以上的类别中心,则将该分子标签剔除。
作为上述方法优选的,标签群内分子标签的搜索是通过前缀查询树实现。
作为上述方法优选的,编辑距离阈值为2~4。
作为上述方法优选的,步骤S2中,质控包括:
S21:剔除捕获片段在预定范围外的读段;
S22:剔除低测序质量的读段;
S23:剔除读段的起始位置和终止位置不在目标区域内的读段;
S24:剔除序列比对质量小于5的读段;
S25:剔除没有比对上的读段。
作为上述方法的优选,步骤S6进一步包括:
S61:在分子标签数目≥3条第二标签群内,分子标签对应读段对应位置的碱基型一致性≥80%,则标记为此碱基型,否则标记为参考基因组对应位置的碱基型,构建成一致性序列;
S62:根将一致性序列与参考基因组比对结果进行突变分析,输出至少有2条一致性序列支持的突变结果。
一种低频突变测序数据分析装置,包括:
数据获取单元:用于获取基于分子标签构建的扩增子文库测序下机数据,所述下机数据包括读段和分子标签;
质控单元:用于根据读段与参考基因组的比对结果进行质控;
第一标签群构建单元:用于将具有相同读段的分子标签集合为第一标签群;
类别中心构建单元:用于在第一标签群内,根据频数和分子标签之间的编辑距离,找出聚类的类别中心;
第二标签群构建单元:用于在第一标签群内,将非类别中心的分子标签归类于相似性最高的类别中心,形成具有相似标签序列的第二标签群;
突变分析单元:用于在第二标签群内,构建一致性序列,根据一致性序列与参考基因组的比对结果进行突变分析,输出突变结果。
作为上述装置优选的,类别中心构建单元进一步包括:
频数统计模块:用于在第一标签群内,统计分子标签的频数;
类别中心识别模块:用于针对频数为L的当前分子标签,找出与当前分子标签编辑距离≤阈值范围的分子标签集合;如果集合内没有序列,则当前分子标签作为一个类别中心;否则,将当前分子标签的频数均分到集合内频数≥5L的分子标签的频数上,当前分子标签的频数则归0;对上述过程进行重复递归,遍历每条分子标签;最后将频数>0的分子标签作为类别中心。
作为上述装置优选的,类别中心构建单元和第二标签群构建单元中,标签群内分子标签的搜索是通过前缀查询树实现。
本发明的有益效果是:
本发明基于分子标签的低频突变测序数据分析方法和装置,是基于对分子标签的频数迭代收敛识别聚类中心,从而实现高效准确的聚类,经分析测试证明,该聚类策略的分析结果相比于普通的聚类模型(基于密度聚类)的分析结果更加准确,能排除PCR扩增过程和测序过程中的出现的错误,进而解决测序分析结果中的背景噪音问题,提高检测的灵敏度和特异度。
附图说明
图1是低频突变测序数据分析方法流程图;
图2是低频突变测序数据分析装置示意图。
具体实施方式
本发明基于分子标签法,发明人在测序文库构建时,引入随机分子标签,随机分子标签长度优选为6~14bp,利用随机分子标签标记初始扩增子,可以识别来源与同一条模板DNA扩增获得的目标扩增子,从而反映真实的突变情况,降低测序错误及复制错误对突变检测结果的干扰。
随机分子标签的引入属于本领域现有技术,技术人员可根据不同的测序平台及文库构建方法选择不同的引入方式,最终获得随机分子标签单端或双端标记的扩增子文库,经高通量测序,获取下机数据,下机数据包括读段(reads)和提取的分子标签序列。
ctDNA突变属于低频突变,本发明以此为例,提供一种低频突变测序数据分析方法和装置,但可以预想本发明的保护范围不限于此,其他基于分子标签的低频突变检测同样适用。
参考图1,本发明提供一种低频突变测序数据分析方法,包括:
S1:获取基于分子标签构建的扩增子文库测序下机数据,所述下机数据包括读段和分子标签;
S2:根据读段与参考基因组的比对结果进行质控;
S3:将具有相同读段的分子标签集合为第一标签群;
S4:在第一标签群内,根据频数和分子标签之间的编辑距离,找出聚类的类别中心;
S5:在第一标签群内,将非类别中心的分子标签归类于相似性最高的类别中心,形成具有相似标签序列的第二标签群;
S6:在第二标签群内,构建一致性序列,根据一致性序列与参考基因组的比对结果进行突变分析,输出突变结果。
作为上述方法优选的,步骤S4进一步包括:
S41:在第一标签群内,统计分子标签的频数;
S42:针对频数为L的当前分子标签,找出与当前分子标签编辑距离≤阈值范围的分子标签集合;如果集合内没有序列,则当前分子标签作为一个类别中心;否则,将当前分子标签的频数均分到集合内频数≥5L的分子标签的频数上,当前分子标签的频数则归0;
S43:对步骤S42进行重复递归,遍历每条分子标签;
S44:将频数>0的分子标签作为类别中心。
作为上述方法优选的,步骤S5进一步包括:
S51:在第一标签群内,找出与非类别中心的分子标签编辑距离最小的类别中心,将所述分子标签归类于所述类别中心,形成具有相似标签序列的第二标签群;
S52:如果同一分子标签归类于两个及以上的类别中心,则将该分子标签剔除。
作为上述方法优选的,标签群内分子标签的搜索是通过前缀查询树实现,基于前缀查询树的算法结构可以实现相似序列的快速搜索,大大提高了分析效率。
作为上述方法优选的,编辑距离阈值为2~4。
作为上述方法优选的,步骤S2中,质控包括:
S21:剔除捕获片段在预定范围外的读段;
S22:剔除低测序质量的读段;
S23:剔除读段的起始位置和终止位置不在目标区域内的读段;
S24:剔除序列比对质量小于5的读段;
S25:剔除没有比对上的读段。
作为上述方法的优选,步骤S6进一步包括:
S61:在分子标签数目≥3条第二标签群内,分子标签对应读段对应位置的碱基型一致性≥80%,则标记为此碱基型,否则标记为参考基因组对应位置的碱基型,构建成一致性序列;
S62:根将一致性序列与参考基因组比对结果进行突变分析,输出至少有2条一致性序列支持的突变结果。
对于上述方法,针对双端标记有分子标签的读段,可将双端分子标签合并为一条分子标签序列进行后续分析,单端标记有分子标签的读段则直接用于分析,双端标记有分子标签的读段可在质控步骤上对分子标签数目进行质控,保留双端均有分子标签的读段,实现尽可能剔除无效读段的质控目的。
质控剔除捕获片段在预定范围外的读段中,所述预定范围根据实际测序捕获情况而定,举例说明,对于探针捕获cfDNA目标片段,一般而言,预定范围为30~400bp,对于多重PCR捕获cfDNA目标片段,一般而言,预定范围为30~180bp。
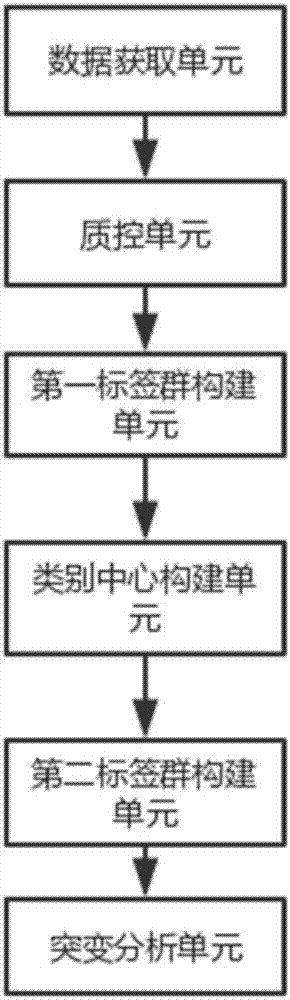
参考图2,基于本发明方法,还提供一种低频突变测序数据分析装置,包括:
数据获取单元:用于获取基于分子标签构建的扩增子文库测序下机数据,所述下机数据包括读段和分子标签;
质控单元:用于根据读段与参考基因组的比对结果进行质控;
第一标签群构建单元:用于将具有相同读段的分子标签集合为第一标签群;
类别中心构建单元:用于在第一标签群内,根据频数和分子标签之间的编辑距离,找出聚类的类别中心;
第二标签群构建单元:用于在第一标签群内,将非类别中心的分子标签归类于相似性最高的类别中心,形成具有相似标签序列的第二标签群;
突变分析单元:用于在第二标签群内,构建一致性序列,根据一致性序列与参考基因组的比对结果进行突变分析,输出突变结果。
作为上述装置优选的,类别中心构建单元进一步包括:
频数统计模块:用于在第一标签群内,统计分子标签的频数;
类别中心识别模块:用于针对频数为L的当前分子标签,找出与当前分子标签编辑距离≤阈值范围的分子标签集合;如果集合内没有序列,则当前分子标签作为一个类别中心;否则,将当前分子标签的频数均分到集合内频数≥5L的分子标签的频数上,当前分子标签的频数则归0;对上述过程进行重复递归,遍历每条分子标签;最后将频数>0的分子标签作为类别中心。
作为上述装置优选的,类别中心构建单元和第二标签群构建单元中,标签群内分子标签的搜索是通过前缀查询树实现。
以下通过具体实施例进一步解释本发明,以下所述仅为本发明的具体实施例,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种变型和改进。在不脱离本发明的设计思路的前提下,所做的任何修改,等同替换等,均应在本发明的保护范围内。
实施例1
本实施例针对已知8个突变位点,突变频率为0.1%~0.13%的样本(肿瘤标准品HD779,来源于horizon discovery)进行ctDNA低频突变测序数据分析。
本实施例双端引入8bp随机分子标签,以cfDNA为模板进行多重PCR捕获,经文库构建、质控、最后利用Proton测序仪进行测序。
低频突变测序数据分析流程如下:
1、数据获取
获取上述分子标签构建的扩增子文库测序下机数据,所述下机数据包括读段和分子标签。
2、质控
用tmap(Life Technologies)软件将读段与人类参考基因组(UCSC数据库的hg19版本)进行比对,根据比对结果进行质控:剔除捕获片段在预定范围(30~180bp)外的读段;剔除Q17碱基比例小于50%(即低测序质量)的读段;剔除读段的起始位置和终止位置不在目标区域内的读段;剔除序列比对质量(mapping quality)小于5的读段;剔除没有比对上的读段;保留两端均有分子标签的读段。
3、第一标签群构建
使用samtools软件的sort工具进行排序,获取每条读段中的起始位置和终止位置,将具有相同起始位置和终止位置的读段分至同一类,将属于同一类的读段的分子标签集合成第一标签群,其中,双端分子标签合并为一条分子标签序列。
4、类别中心构建
在第一标签群内,统计分子标签的频数;针对频数为L的当前分子标签,利用前缀查询树找出与当前分子标签编辑距离≤2的分子标签集合;如果集合内没有序列,则当前分子标签作为一个类别中心;否则,将当前分子标签的频数均分到集合内频数≥5L的分子标签的频数上,当前分子标签的频数则归0;对上述过程进行重复递归,遍历每条分子标签;最后将频数>0的分子标签作为类别中心。
5、第二标签群构建
在第一标签群内,利用前缀查询树找出与非类别中心的分子标签编辑距离最小的类别中心,将所述分子标签归类于所述类别中心,形成具有相似标签序列的第二标签群;如果同一分子标签归类于两个及以上的类别中心,则将该分子标签剔除。
6、构建一致性序列
在分子标签数目≥3条第二标签群内,提取分子标签对应的读段,读段内对应位置的碱基型一致性≥80%,则标记对应位置的碱基为此碱基型,否则标记为人类参考基因组(hg19)序列对应位置的碱基型,最终构建成一致性序列。
7、校正
用tmap软件将一致性序列比对至人类参考基因组(hg19),使用samtools软件的mpileup工具提取突变信息,对于一致性序列检出的突变信息,至少有2条一致性序列支持的突变结果作为阳性检出。
表1给出5M数据量下,本实施例及常规聚类分析获得的检测结果,其中,常规聚类分析是指在获得第一标签群的基础上采用密度聚类分析获得第二标签群,其他方法同本实施例。由表可见,8个0.1%水平的突变中能够成功检出8个突变;在常规聚类分析测试中,有2个点未检出。
表1、实施例1与常规方法的比较
注:分子深度是指覆盖该点的一致性序列深度,实测频率是指支持突变的一致性序列深度/分子深度,测序深度是指覆盖该点的原始测序深度。
实施例2
本实施例针对已知8个突变位点,突变频率为1%~1.3%的样本(肿瘤标准品HD778,来源于horizon discovery)进行ctDNA低频突变测序数据分析。本实施例的其他检测流程同
实施例1,不在此不一一赘述。
表2给出1M数据量下,本实施例及常规聚类分析获得的检测结果,其中,常规聚类分析是指在获得第一标签群的基础上采用密度聚类分析获得第二标签群,其他方法同实施例1。由表可见,本实施例检测1%标准品灵敏度100%和特异度100%;常规聚类分析测试结果一致。
表2、实施例2与常规方法的比较
注:分子深度是指覆盖该点的一致性序列深度,实测频率是指支持突变的一致性序列深度/分子深度,测序深度是指覆盖该点的原始测序深度。
- 还没有人留言评论。精彩留言会获得点赞!