一种氢氧燃料电池阳极铱基催化剂的制备方法与流程
本发明属于燃料电池技术领域,特别涉及一种氢氧燃料电池阳极铱基催化剂的制备方法。
背景技术:
氢氧燃料电池是一种将氢气中的化学能直接转换为电能的装置,它具有能量密度高、功率密度高、能量转换效率高、无污染、室温快速启动等突出特点,被认为是未来电动汽车的最佳动力电池。然而,氢氧燃料电池中使用大量贵金属铂作为催化剂,导致其成本居高不下,制约了其大规模商业化推广。开发氢氧燃料电池阳极非铂催化剂,对降低燃料电池成本、实现燃料电池汽车大规模商业化具有重要意义。
金属铱催化氢氧化反应具有过电位低、反应速率较高的特点,被认为是潜在的铂替代催化剂之一,备受研究者的关注。文献[int.j.hydrogen.energy,2010,35:5528-5538]公开了一种通过水热还原合成irv/c催化剂的方法,该方法首先采用120℃乙二醇溶剂热还原ir、v前驱体,然后在200℃还原气氛退火获得irv/c催化剂。文献[j.phys.chem.c,2011,115:9894-9902]报道了一种irni/c核壳结构催化剂的合成方法,该方法采用nabh4还原剂金属前驱体,再在还原气氛600℃高温处理,获得irni/c核壳结构催化剂。中国发明专利cn101411012a公开了“一种燃料电池用催化剂的制造方法”,该发明通过ph值调节,使各金属盐形成氢氧化物担载在导电载体上,再通过两步热处理合金化,制备出含铂、贱金属和铱的三元合金催化剂。文献[j.mater.chem.a,2014,2:10098-10103]和中国发明专利cn103331172a公开了“一种燃料电池ir基阳极催化粒径剂的制造方法”,该方法先以浓氨水为络合剂形成镍氨络合阳离子,再水浴蒸干制备出碳载铱镍络合物,然后在氢气氛中还原,制备出了碳载irni合金催化剂。以上方法制备出的铱基催化剂虽然皆具有一定的氢氧化活性,但催化性能都仍难以与铂基催化剂相媲美,催化活性无法满足燃料电池实际应用的要求;且制备工艺都复杂、繁琐、污染大,难以实现催化剂的绿色、批量化制备。
技术实现要素:
本发明的目的是针对现有的铱基催化剂制备方法工艺繁琐、成本高、无法批量生产且活性不足的问题,提供一种氢氧燃料电池阳极铱基催化剂的制备方法。本发明先将铱前驱体、功能化碳载体以及柠檬酸钠均匀分散在溶剂中,搅拌一段时间后,将该混合液均匀地分布在有机聚合物薄膜上,再水浴蒸干;最后通过在还原气氛热处理,制备出颗粒均匀分散的碳载铱基催化剂。本发明大大简化了传统的铱基催化剂制备方法,所制备的催化剂活性高、产量大、一致性好、粒径小、高分散,使得金属利用率及氢氧化活性得到有效提高,并且能够批量生产。
本发明的目的是这样实现的:一种氢氧燃料电池阳极铱基催化剂的制备方法,其具体方法步骤包括
(1)碳载体的功能化
取500ml圆底烧瓶,称取8克市售vulcanxc-72碳粉,加入100~320毫升浓硝酸后,升温回流至沸腾开始计时,反应2~8h,冷却至室温,加水稀释,抽滤洗涤至ph接近7,烘干,球磨成粉末,即得到功能化vulcanxc-72碳粉。
(2)铱前驱体吸附在功能化碳上
按功能化vulcanxc-72碳粉∶铱∶柠檬酸钠的质量比1∶0.05~2∶0.1~12分别称取步骤(1)所得功能化vulcanxc-72碳粉、铱前驱体和柠檬酸钠;先将功能化vulcanxc-72碳粉加入去离子水中,超声分散10~60分钟,形成均匀分散的功能化vulcanxc-72碳粉混合液,其中功能化vulcanxc-72碳粉的质量浓度为3~100毫克/毫升;然后依次加入铱前驱体、柠檬酸钠,搅拌2~24小时,接着,将所得液体转移到平铺的有机聚合物薄膜上,再在50~80℃下水浴蒸干、研磨成粉。
其中所述铱前驱体为氯铱酸、氯铱酸钠、氯亚铱酸钠的其中之一。
(3)碳载铱基催化剂的制备
将上述所得的粉末在还原性气体∶惰性气体的体积比为1∶1~1∶20,气体总流量为50~200毫升/分钟的混合气氛下200~800℃热处理1~3小时,在混合气氛下自然冷却。最后将批量制备出的上述粉末分散在50~500毫升去离子水中后,超声洗涤5~30分钟,再搅拌洗涤1~4小时,接着过滤洗涤3~10次、真空干燥得到碳载铱基催化剂。
本发明采用上述技术方案后,主要有以下优点:
(1)本发明利用在平铺的有机聚合物薄膜上水浴蒸干,克服了水浴蒸干时间长、耗能大的缺点,在充分发挥柠檬酸钠控制形成的颗粒粒径作用的同时,更是解决了加入柠檬酸钠所造成的蒸干产物极难脱取、损失大、易潮解等难题,也解决了在批量生产中化学成分一致性差、难以均匀分散的难题,从而得到产率高、化学成分均一且分布均匀的超细纳米粒子,金属利用率及催化剂活性大大提高。
(2)无需合金化、无需水热还原步骤、无需ph值调控步骤、无需离心洗涤步骤,直接以还原气退火处理水浴蒸干产物合成催化剂,最后简单的清洗即可获得表面清洁、高分散、高活性的催化剂,操作简单易行、工艺环保经济。
(3)利用该方法批量制备出的催化剂与微量制备的催化剂在活性上没有差别,并且前后纯化工艺简单易行,所以该方法适用于商业化、低成本批量生产铱基催化剂。
(4)相比于现有的商业化铂基催化剂,该方法制备的碳载铱基催化剂具有能相媲美的高活性,且具有明显的成本优势,可以有效降低燃料电池成本。
本发明方法简单易行,操作安全,生产成本低廉,产品性能优异,非常适用于批量生产。采用本发明制备的碳载铱基催化剂具有高效的催化氢氧化性能,可替代铂基催化剂应用于燃料电池领域,特别是质子交换膜燃料电池氢阳极,并能实现催化剂的大规模商业化生产。
附图说明
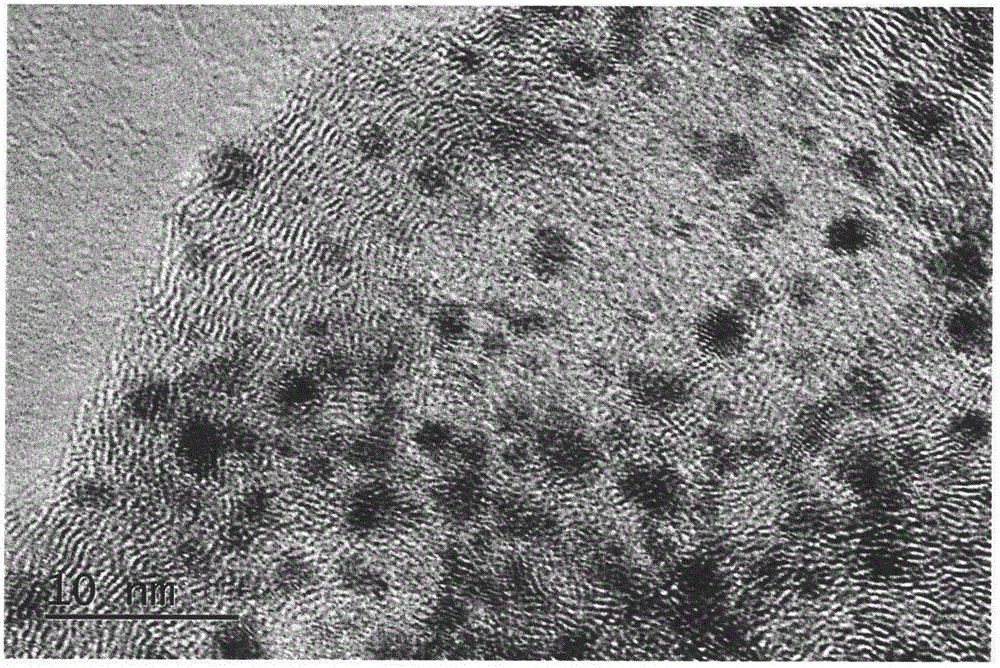
图1为实施例1制得的铱碳催化剂的高分辨透射电子显微镜(hrtem)图。
图2为实施例1制得的铱碳催化剂和以对比实验1的英国jonhson-matthey公司商业化pt/c(铂质量百分比20%)催化剂的氢氧化线性扫描曲线图。曲线a是以实施例1制备的铱碳催化剂为工作电极,银/氯化银电极为参比电极,铂环为对电极,氢气饱和的0.1摩尔/升高氯酸水溶液为电解液,扫描速度为10毫伏/秒条件下的的氢氧化线性扫描曲线。曲线b是以英国jonhson-matthey公司商业化pt/c催化剂为工作电极,银/氯化银电极为参比电极,铂环为对电极,氢气饱和0.1摩尔/升高氯酸水溶液为电解液,扫描速度为10毫伏/秒条件下的的氢氧化线性扫描曲线。其中工作电极上铱载量与铂载量均为5微克,电极旋转速度均为1600转/分钟。
图3为实施例1制得的铱碳催化剂和以对比实验2的英国jonhson-matthey公司商业化ptru/c(铂质量百分比20%)催化剂的氢氧化线性扫描曲线图。曲线a是以实施例1制备的铱碳催化剂为工作电极,银/氯化银电极为参比电极,铂环为对电极,氢气饱和的0.1摩尔/升氢氧化钾水溶液为电解液,扫描速度为10毫伏/秒条件下的的氢氧化线性扫描曲线。曲线b是以英国jonhson-matthey公司商业化ptru/c催化剂为工作电极,银/氯化银电极为参比电极,铂环为对电极,氢气饱和0.1摩尔/升氢氧化钾水溶液为电解液,扫描速度为10毫伏/秒条件下的的氢氧化线性扫描曲线。其中工作电极上铂钌总载量与铱载量均为5微克,电极旋转速度均为1600转/分钟。
图4为实施例1、实施例2制备的铱碳催化剂的氢氧化线性扫描曲线图。曲线a是以实施例2制备的铱碳催化剂为工作电极,银/氯化银电极为参比电极,铂环为对电极,氢气饱和的0.1摩尔/升氢氧化钾水溶液为电解液,扫描速度为10毫伏/秒条件下的的氢氧化线性扫描曲线。曲线b是以实施例1制备的铱碳催化剂为工作电极,银/氯化银电极为参比电极,铂环为对电极,氢气饱和的0.1摩尔/升氢氧化钾水溶液为电解液,扫描速度为10毫伏/秒条件下的的氢氧化线性扫描曲线。其中工作电极上铱载量均为5微克,电极旋转速度均为1600转/分钟。
图5为实施例1制得的铱碳催化剂和以对比实验3的碳载铱镍合金催化剂的氢氧化线性扫描曲线图。曲线a是以实施例1制备的铱碳催化剂为工作电极,银/氯化银电极为参比电极,铂环为对电极,氢气饱和的0.1摩尔/升氢氧化钾水溶液为电解液,扫描速度为10毫伏/秒条件下的的氢氧化线性扫描曲线。曲线b是对比实验3的碳载铱镍合金催化剂为工作电极,银/氯化银电极为参比电极,铂环为对电极,氢气饱和0.1摩尔/升氢氧化钾水溶液为电解液,扫描速度为10毫伏/秒条件下的的氢氧化线性扫描曲线。其中工作电极上铂钌总载量与铱载量均为5微克,电极旋转速度均为1600转/分钟。
具体实施方式
下面结合具体实施方式,进一步说明本发明。
实施例1
(1)碳载体的功能化
取500ml圆底烧瓶,称取8克市售vulcanxc-72碳粉,加入320毫升浓硝酸后,升温回流至沸腾开始计时,反应4.5h,冷却至室温,加水稀释,抽滤洗涤至ph接近7,烘干,球磨成粉末,即得到功能化vulcanxc-72碳粉。
(2)铱前驱体吸附在功能化碳上
按功能化vulcanxc-72碳粉∶铱∶柠檬酸钠的质量比1∶0.33∶2分别称取步骤(1)所得功能化vulcanxc-72碳粉、铱前驱体和柠檬酸钠;先将功能化vulcanxc-72碳粉(8克)加入去离子水中,超声分散30分钟,形成均匀分散的功能化vulcanxc-72碳粉混合液,其中功能化vulcanxc-72碳粉的质量浓度为100毫克/毫升;然后依次加入铱前驱体、柠檬酸钠,搅拌24小时,接着,将所得液体转移到平铺的有机聚合物薄膜上,再在72℃下水浴蒸干、研磨成粉。
(3)铱碳催化剂的制备
将上述所得的粉末在氢气∶氮气的体积比为1∶6,气体总流量为70毫升/分钟的混合气氛下500℃热处理2小时,在混合气氛下自然冷却。最后将批量制备出的上述粉末分散在150毫升去离子水中后,超声洗涤10分钟,再搅拌洗涤4小时,接着过滤洗涤4次、真空干燥得到铱碳催化剂。
(4)铱碳催化剂的透射电子显微镜测试
制备好的铱碳催化剂用透射电镜测试得到图1中的高分辨透射电镜(hrtem)照片。
(5)催化剂在三电极体系的电化学性能测试
称取1毫克第(4)步所制得的铱碳催化剂加入到1000微升无水乙醇中,超声振荡30分钟分散均匀后,微量进样器吸取20微升均匀涂覆于玻璃碳旋转圆盘电极上,60℃下保持10分钟。以此为工作电极,以铂环电极和银/氯化银(ag/agcl)电极分别作为辅助电极和参比电极。
在氮气饱和的0.1摩尔/升的高氯酸溶液中循环伏安扫描35圈以活化催化剂。扫描速率为50毫伏/秒,扫描范围为0~0.9伏(相对于标准氢电极)。对催化剂进行表面活化后,在氢气饱和的0.1mol/l的高氯酸溶液中测试线形扫描伏安曲线,旋转电极的转速为1600转/分钟,扫描速率10毫伏/秒,扫描范围-0.01~0.3伏(相对于标准氢电极),氢氧化线性扫描结果对应图2中曲线a。
在氮气饱和的0.1摩尔/升的氢氧化钾溶液中循环伏安扫描35圈以活化催化剂。扫描速率为50毫伏/秒,扫描范围为0~0.9伏(相对于标准氢电极)。对催化剂进行表面活化后,在氢气饱和的0.1mol/l的氢氧化钾溶液中测试线形扫描伏安曲线,旋转电极的转速为1600转/分钟,扫描速率10毫伏/秒,扫描范围-0.01~0.3伏(相对于标准氢电极),氢氧化线性扫描结果对应图3中曲线a、图4中曲线b和图5中曲线a。
实施例2
步骤(1)同实施例1中步骤(1)。
(2)铱前驱体吸附在功能化碳上
按功能化vulcanxc-72碳粉∶铱∶柠檬酸钠的质量比1∶0.25∶1.5分别称取步骤(1)所得功能化vulcanxc-72碳粉、铱前驱体和柠檬酸钠;先将功能化vulcanxc-72碳粉(0.16克)加入去离子水中,超声分散30分钟,形成均匀分散的功能化vulcanxc-72碳粉混合液,其中功能化vulcanxc-72碳粉的质量浓度为4毫克/毫升;然后依次加入铱前驱体、柠檬酸钠,搅拌24小时,接着,将所得液体转移到平铺的有机聚合物薄膜上,再在72℃下水浴蒸干、研磨成粉。
步骤(3)同实施例1中步骤(3)。
(4)催化剂在三电极体系的电化学性能测试
称取2毫克第(4)步所制得的铱碳催化剂加入到1000微升无水乙醇中,超声振荡30分钟分散均匀后,微量进样器吸取12.5微升均匀涂覆于玻璃碳旋转圆盘电极上,60℃下保持10分钟。以此为工作电极,以铂环电极和银/氯化银(ag/agcl)电极分别作为辅助电极和参比电极。在氮气饱和的0.1摩尔/升的氢氧化钾溶液中循环伏安扫描35圈以活化催化剂。扫描速率为50毫伏/秒,扫描范围为0~0.9伏(相对于标准氢电极)。对催化剂进行表面活化后,在氢气饱和的0.1mol/l的氢氧化钾溶液中测试线形扫描伏安曲线,旋转电极的转速为1600转/分钟,扫描速率10毫伏/秒,扫描范围-0.01~0.3伏(相对于标准氢电极),氢氧化线性扫描结果对应图4中曲线a。
对比实验1
英国jonhson-matthey公司商业化pt/c(铂质量百分比20%)催化剂在三电极体系的电化学性能测试电化学测试催化剂在三电极体系的电化学性能测试
称取2毫克商业化pt/c催化剂加入到1000微升无水乙醇中,超声振荡10分钟分散均匀后,微量进样器吸取12.5微升均匀涂覆于玻璃碳旋转圆盘电极上,60℃下保持10分钟。以此为工作电极,以铂环电极和银/氯化银(ag/agcl)电极分别作为辅助电极和参比电极。在氮气饱和的0.1摩尔/升的高氯酸溶液中循环伏安扫描35圈以活化催化剂。扫描速率为50毫伏/秒,扫描范围为0~0.9伏(相对于标准氢电极)。对催化剂进行表面活化后,在氢气饱和的0.1mol/l的高氯酸溶液中测试线形扫描伏安曲线,旋转电极的转速为1600转/分钟,扫描速率10毫伏/秒,扫描范围-0.01~0.3伏(相对于标准氢电极),氢氧化线性扫描结果对应图2中曲线b。
对比实验2
英国jonhson-matthey公司商业化ptru/c(铂钌质量分数为20%)催化剂在三电极体系的电化学性能测试电化学测试
称取2毫克商业化ptru/c催化剂加入到1000微升无水乙醇中,超声振荡10分钟分散均匀后,微量进样器吸取12.5微升均匀涂覆于玻璃碳旋转圆盘电极上,60℃下保持10分钟。以此为工作电极,以铂环电极和银/氯化银(ag/agcl)电极分别作为辅助电极和参比电极。在氮气饱和的0.1摩尔/升的氢氧化钾溶液中循环伏安扫描35圈以活化催化剂。扫描速率为50毫伏/秒,扫描范围为0~0.9伏(相对于标准氢电极)。对催化剂进行表面活化后,在氢气饱和的0.1mol/l的氢氧化钾溶液中测试线形扫描伏安曲线,旋转电极的转速为1600转/分钟,扫描速率10毫伏/秒,扫描范围-0.01~0.3伏(相对于标准氢电极),氢氧化线性扫描结果对应图3中曲线b。
对比实验3
按照文献[j.mater.chem.a,2014,2:10098-10103]和中国发明专利cn103331172a公开的“一种燃料电池ir基阳极催化剂的制造方法”,制备出了碳载irni合金催化剂:
(1)碳载体的功能化
取500ml圆底烧瓶,称取1克市售vulcanxc-72碳粉,加入30%过氧化氢∶浓硫酸体积比为1∶4的混合溶液150毫升,超声并搅拌3小时后,用超纯水稀释,静置24小时后滤出上层清液,经多次离心洗涤,烘干,研磨后得到功能化vulcanxc-72碳粉。
(2)碳载铱镍络合物的制备
按功能化vulcanxc-72碳粉∶铱∶镍∶柠檬酸钠的质量比1∶0.3∶0.2∶1.8分别称取步骤(1)所得功能化vulcanxc-72碳粉、氯铱酸、氯化镍和柠檬酸钠;先将功能化vulcanxc-72碳粉加入去离子水中,超声分散30分钟,形成均匀分散的质量浓度为8毫克/毫升的功能化vulcanxc-72碳粉悬浮液;然后依次加入氯铱酸、氯化镍和柠檬酸钠,先超声分散20分钟,继续搅拌12小时,再用质量浓度为28%的氨水调节ph值至12,密封搅拌18小时,然后在60℃水浴烘干、研磨成粉,得到碳载铱镍络合物。
(3)碳载铱镍合金催化剂的制备
将步骤(2)所得的碳载铱镍络合物在氢气∶氩气体积比为1∶6,气体总流量为350毫升/分钟的混合气氛下500℃热处理2小时,然后在混合气氛下自然冷却,最后水洗、真空干燥得到碳载铱镍合金催化剂。
(4)催化剂在三电极体系的电化学性能测试
称取2毫克第(3)步所制得的碳载铱镍合金催化剂加入到1000微升无水乙醇中,超声振荡10分钟分散均匀后,微量进样器吸取12.5微升均匀涂覆于玻璃碳旋转圆盘电极上,60℃下保持10分钟。以此为工作电极,以铂环电极和银/氯化银(ag/agcl)电极分别作为辅助电极和参比电极,在氮气饱和的0.1摩尔/升的氢氧化剂水溶液中循环伏安扫描35圈。扫描速率为50毫伏/秒,扫描范围为0~0.9伏(相对于标准氢电极)。对催化剂进行表面活化后,在氢气饱和的0.1mol/l的氢氧化钾溶液中测试线形扫描伏安曲线,旋转电极的转速为1600转/分钟,扫描速率10毫伏/秒,扫描范围-0.01~0.3伏(相对于标准氢电极),氢氧化线性扫描结果对应图5中曲线b。
本发明的试验结果:
从图1可以看出,本发明所制备的铱碳催化剂颗粒小,高分散,一致性好。
从图2可以看出,在贵金属载量相同的情况下,在酸性介质中进行的氢氧化线性扫描测试中,本发明所制备的铱碳催化剂与英国jonhson-matthey公司商业化pt/c催化剂相比,塔菲尔斜率有所增加,极限电流可与pt/c相媲美,表明本发明制备的铱碳催化剂在酸性介质中的氢氧化的催化活性要优于jonhson-matthey公司商业化pt/c催化剂。
从图3可以看出,在贵金属载量相同的情况下,本发明所制备的铱碳催化剂与英国jonhson-matthey公司商业化ptru/c催化剂相比,在碱性电解质中的塔菲尔斜率、极限电流均显著增加,表明本发明制备的催化剂在碱性介质中的氢氧化的催化活性要显著优于jonhson-matthey公司商业化ptru/c催化剂。
从图4可以看出,本发明所批量制备的催化剂和微量制备的催化剂在氢氧化线性扫描测试中,表现出几乎完全相同的氢氧化催化活性,表现该方法适用于批量生产铱基催化剂。
从图5可以看出,本发明所批量制备的催化剂,与文献[j.mater.chem.a,2014,2:10098-101031和中国发明专利cn103331172a所公开的方法制备出的微量催化剂相比较,表现出显著优异的氢氧化的催化活性。
- 还没有人留言评论。精彩留言会获得点赞!