妥布赖森化合物、制备和使用方法与流程
本发明涉及在结构上与妥布赖森(tubulysins)类似的化合物、其与配体的缀合物、制备和使用此类化合物和缀合物的方法和包含此类化合物和缀合物的组合物。
妥布赖森是最初从粘细菌:过渡型原囊粘细菌(Archangium gephyra)或碟形囊球粘细菌(Angiococcus disciformis)的培养物分离的细胞毒素,其中各生物体产生妥布赖森的不同混合物(Sasse等人,2000;Reichenbach等人,1998)。已阐明其晶体结构和生物合成途径(Steinmetz等人,2004)且已对其生物合成基因进行测序(Hoefle等人,2006b)。前妥布赖森(pretubulysin),妥布赖森的生物合成前体,也已显示拥有一些活性(Ullrich等人,2009)。(本文通过第一作者或发明人和年份引用的文献的全部引文都列于本说明书结尾处)。
妥布赖森属于天然存在的抗有丝分裂多肽和缩肽的组群,该组群包括拟茎点霉毒素(phomopsin)、尾海兔素(dolastatin)和念珠藻素(cryptophycin) (Hamel 2002)。还存在除多肽或缩肽外的抗有丝分裂剂,例如紫杉醇(paclitaxel)、美坦辛(maytansine)和埃博霉素(epothilone)。在有丝分裂期间,细胞的微管重新组织以形成有丝分裂纺锤体,这是需要微管组成蛋白α-微管蛋白和β-微管蛋白的快速装配和去装配(disassembly)的过程。抗有丝分裂剂阻断该过程并阻止细胞发生有丝分裂。在分子水平上,一种抗有丝分裂剂的确切阻断机制可以与另一种不同。妥布赖森防止微管蛋白装配成微管,从而使得所影响细胞累积于G2/M期中并发生细胞凋亡(Khalil等人,2006)。紫杉醇通过结合至微管并防止其去装配来实现相同最终结果。
妥布赖森具有从如式(A)中所显示的一个蛋白氨基酸亚单元和三个非蛋白氨基酸亚单元构成的四肽基支架:N-甲基哌啶酸(Mep)、异亮氨酸(Ile)、管式缬氨酸(Tuv,tubuvaline)和管式苯丙氨酸(Tup,tubuphenylalanine,R'等于H)或管式酪氨酸(Tut,tubutyrosine,R'等于OH)。在更为熟知的天然存在的妥布赖森(称为A、B等)中,结构变化的位点位于式(A)的残基R'、R''和R'''处,如表1中所显示:
此外,已鉴定出其它天然存在的妥布赖森(Chai等人,2010)。
Kaur等人,2006研究了妥布赖森A的抗增殖性质且发现,其比其它抗有丝分裂剂(例如紫杉醇和长春碱(vinblastine))更有效且在针对各种癌细胞系的异种移植物测定中具有活性。此外,妥布赖森A诱导癌细胞但非正常细胞中的细胞凋亡且在体外测定中显示显著潜在抗血管生成性质。也已评估其它妥布赖森的抗有丝分裂性质且通常发现可有利地与非妥布赖森抗有丝分裂剂的性质进行比较(参见例如Balasubramanian等人,2009;Steinmetz等人,2004;Wipf等人,2004)。出于这些原因,极为关注使用妥布赖森作为抗癌剂(参见例如Domling等人,2005c;Hamel 2002)。
许多出版物描述了针对合成妥布赖森的成果,包括:Balasubramanian等人,2009;Domling等人,2006;Hoefle等人,2003;Neri等人,2006;Peltier等人,2006;Sani等人,2007;Sasse等人,2007;Shankar等人,2009;Shibue等人,2009和2010;和Wipf等人,2004。其它出版物描述了经由制备和评估妥布赖森类似物或衍生物的结构-活性关系(SAR)研究:Balasubramanian等人,2008和2009;Chai等人,2011;Domling 2006;Domling等人,2005a;Ellman等人,2013;Hoefle等人,2001和2006a;Pando等人,2011;Patterson等人,2007和2008;Richter,2012a、2012b和2012c;Shankar等人,2013;Shibue等人,2011;Sreejith等人,2011;Vlahov等人,2010a;Wang等人,2007;Wipf等人,2007和2010;和Zanda等人,2013。SAR研究主要探究Mep环、Tuv亚单元的残基R''和R'''和Tup/Tut亚单元的芳族环或脂族碳链中的结构变化。
Domling等人,2005公开妥布赖森与配偶体分子的缀合物,配偶体分子一般描述为聚合物或生物分子,但实例限于聚乙二醇(PEG)作为配偶体分子。Cheng等人,2011也公开适用于缀合物中的妥布赖森类似物。其它公开妥布赖森缀合物的文献是Boyd等人,2008和2010;Jackson等人,2013;Vlahov等人,2008a、2008b和2010b;Leamon等人,2008和2010;Reddy等人,2009;和Low等人,2010。Leung等人2002公开可缀合至药物(包括妥布赖森)以改善其生物活性和水溶性的多阴离子多肽。
Davis等人2008和Schluep等人2009公开了基于环糊精的制剂,其中妥布赖森经由键合至Tup/Tut羧基的酰肼-二硫化物接头部分共价连接至环糊精。
据报导,Tuv亚单元的去乙酰化(即式(A)中的R''是羟基而非乙酰基)导致生物活性的损失(Domling等人,2006)。在妥布赖森U和V (其不同之处在于前者被乙酰化而后者被去乙酰化)的研究中,取决于测定,妥布赖森V被报导为效力低约1/600至1/200 (Balasubramanian等人,2009)。因为乙酸酯基团易于水解,所以关注R''位置处的去乙酰化作为导致损失活性的潜在不稳定中心以用于开发用于药物应用的妥布赖森类似物。
发明概述
本发明人已发现,可通过用氨基甲酸酯基团代替R''位置处的乙酸酯来预防如上文讨论与去乙酰化有关的生物活性损失。如本文所述的氨基甲酸酯基团不导致生物活性的显著损失,但仍是更稳定的。
因此,在一方面,本发明提供具有由式(I)代表的结构的化合物:
其中
R1是H、未取代或取代的C1-C10烷基、未取代或取代的C2-C10烯基、未取代或取代的C2-C10炔基、未取代或取代的芳基、未取代或取代的杂芳基、未取代或取代的(CH2)1-2O(C1-C10烷基)、未取代或取代的(CH2)1-2O(C2-C10烯基)、未取代或取代的(CH2)1-2O(C2-C10炔基)、(CH2)1-2OC(=O)(C1-C10烷基)、未取代或取代的(CH2)1-2OC(=O)(C2-C10烯基)、未取代或取代的(CH2)1-2OC(=O)(C2-C10炔基)、未取代或取代的C(=O)(C1-C10烷基)、未取代或取代的C(=O)(C2-C10烯基)、未取代或取代的C(=O)(C2-C10炔基)、未取代或取代的环脂族基团、未取代或取代的杂环脂族基团、未取代或取代的芳基烷基或未取代或取代的烷基芳基;
R2是H、未取代或取代的C1-C10烷基、未取代或取代的C2-C10烯基、未取代或取代的C2-C10炔基、未取代或取代的芳基、未取代或取代的杂芳基、未取代或取代的(CH2)1-2O(C1-C10烷基)、未取代或取代的(CH2)1-2O(C2-C10烯基)、未取代或取代的(CH2)1-2O(C2-C10炔基)、(CH2)1-2OC(=O)(C1-C10烷基)、未取代或取代的(CH2)1-2OC(=O)(C2-C10烯基)、未取代或取代的(CH2)1-2OC(=O)(C2-C10炔基)、未取代或取代的C(=O)(C1-C10烷基)、未取代或取代的C(=O)(C2-C10烯基)、未取代或取代的C(=O)(C2-C10炔基)、未取代或取代的环脂族基团、未取代或取代的杂环脂族基团、未取代或取代的芳基烷基、未取代或取代的烷基芳基或;
其中每个R2a独立地是H、NH2、NHMe、Cl、F、Me、Et或CN;
R3a和R3b独立地是H、C1-C5烷基、CH2(C5-C6环烷基)、CH2C6H5、C6H5或CH2CH2OH;
R4是
、、或
;
其中R4a是H或C1-C3烷基;且Y是H、OH、Cl、F、CN、Me、Et、NO2或NH2;
R5是H、C1-C5烷基、C2-C5烯基、C2-C5炔基、CO(C1-C5烷基)、CO(C2-C5烯基)或CO(C2-C5炔基);
W是O或S (优选地O);且
n是0、1或2;
或其药学上可接受的盐。
在另一个实施方案中,本发明提供缀合物,其包含共价连接至特异性或优先结合至靶细胞上的化学实体的靶向部分的根据式(I)的化合物,该靶细胞优选地是癌细胞。优选地,靶向部分是抗体-更优选地单克隆抗体和甚至更优选地人单克隆抗体-或其抗原结合部分且化学实体是肿瘤相关性抗原。肿瘤相关性抗原可以是展示于癌细胞表面上的抗原或由癌细胞分泌至周围细胞外空间中的抗原。
在另一个实施方案中,提供物质组合物,其包含本发明化合物和具有适于缀合至靶向部分的反应性官能团的接头部分。
在另一个实施方案中,提供治疗患有癌症的受试者的此类癌症的方法,其包括向受试者施用治疗有效量的本发明化合物或其与靶向部分(尤其抗体)的缀合物。在另一个实施方案中,提供本发明化合物或其与靶向部分(尤其抗体)的缀合物用于制备药物的用途,所述药物用于治疗患有癌症的受试者的此类癌症。癌症可以是肾癌、胃癌、肺癌或卵巢癌。
附图简述
图1、2a-2b和3组合显示用于合成化合物(III-1)的方案。
图4显示用于合成化合物(III-2)的方案。
图5显示用于合成化合物(I-2)和(I-3)的方案。
图6a-6c组合显示用于合成化合物(III-4)和(III-5)的方案。
图7显示用于合成化合物(I-1)的方案。
图8显示用于合成化合物(I-4)的方案。
图9a和9b组合显示用于合成化合物(III-6)的方案。
图10和11显示用于合成可用于制备本发明化合物的中间体的方案。
图12显示用于合成其它本发明化合物的方案。
图13a-13d显示一些本发明化合物的生物活性。
图14显示本发明缀合物的体外活性。
图15显示本发明缀合物的体内活性。
图16a、16b、17a、17b、18a、18b、19a、19b和20呈现关于本发明缀合物的活性的其它体内数据。
图21和22显示用于合成其它本发明化合物的方案,其图解说明在氨基甲酸酯基团处的结构变化。
图23显示用于合成适于经由“点击”化学缀合的本发明化合物的方案。
图24显示用于合成适于经由脂族胺基团缀合的本发明化合物的方案。
发明详述
定义
“抗体”意指完整抗体和其任何抗原结合片段(即“抗原结合部分”)或单链变体。完整抗体是包含由二硫键相互连结的至少两条重(H)链和两条轻(L)链的蛋白。每条重链包含重链可变区(VH)和包含三个结构域(CH1、CH2和CH3)的重链恒定区。每条轻链包含轻链可变区(VL或VK)和包含一个单一结构域CL的轻链恒定区。VH区和VL区可进一步细分成超变区(称为互补决定区(CDR)),其被更保守的框架区(FR)穿插。各VH和VL包含3个CDR和4个FR,其从氨基末端至羧基末端以下列顺序排列:FR1、CDR1、FR2、CDR2、FR3、CDR3和FR4。可变区含有与抗原相互作用的结合结构域。恒定区可介导抗体与宿主组织或因子的结合,所述组织或因子包括免疫系统的各种细胞(例如效应细胞)和经典补体系统的第一组分(Clq)。如果抗体以下列KD结合至抗原X,则该抗体被认为“特异性结合”至抗原X:5 × 10-8 M或更小、更优选地1 × 10-8 M或更小、更优选地6 × 10-9 M或更小、更优选地3 × 10-9 M或更小、甚至更优选地2 × 10-9 M或更小。抗体可以是嵌合抗体、人源化抗体或优选地人抗体。重链恒定区可工程改造以影响糖基化类型或程度,延长抗体半衰期,增强或降低与效应细胞或补体系统的相互作用,或调节一些其它性质。可通过以下方式实现工程改造:代替、添加或缺失一个或多个氨基酸或使用来自另一免疫球蛋白类型的结构域代替结构域,或上述方式的组合。
抗体的“抗原结合片段”和“抗原结合部分”(或简单地“抗体部分”或“抗体片段”)意指抗体的一个或多个保持特异性结合至抗原的能力的片段。已显示,抗体的抗原结合功能可通过全长抗体的片段实施,例如(i) Fab片段,即由VL、VH、CL和CH1结构域组成的单价片段;(ii) F(ab')2片段,即包含两个于铰链区处通过二硫桥键连接的Fab片段的二价片段;(iii) Fab'片段,其基本上是具有铰链区的一部分的Fab (参见例如Abbas等人,Cellular and Molecular Immunology,第6版,Saunders Elsevier 2007);(iv) Fd片段,其由VH和CH1结构域组成;(v) Fv片段,其由抗体单臂的VL和VH结构域组成;(vi) dAb片段(Ward等人(1989) Nature 341:544-546),其由VH结构域组成;(vii)分离的互补决定区(CDR);和(viii)纳米抗体,即含有单一可变结构域和两个恒定结构域的重链可变区。优选抗原结合片段是Fab、F(ab')2、Fab'、Fv和Fd片段。此外,尽管Fv片段的两个结构域VL和VH由单独基因编码,但其可使用重组方法通过合成接头来连接,该合成接头能够使其作为蛋白单链(称为单链Fv或scFv)制备,其中VL区与VH区配对以形成单价分子;参见例如Bird等人(1988) Science 242:423-426;和Huston等人(1988) Proc. Natl. Acad. Sci. USA 85:5879-5883)。此类单链抗体还涵盖于术语抗体的“抗原结合部分”内。
“分离抗体”意指实质上不含具有不同抗原特异性的其它抗体的抗体(例如特异性结合抗原X的分离抗体实质上不含特异性结合除抗原X外的抗原的抗体)。然而,特异性结合抗原X的分离抗体可与其它抗原(例如来自其它物种的抗原X分子)具有交叉反应性。在某些实施方案中,分离抗体特异性结合至人抗原X且不与其它(非人)抗原X抗原交叉反应。此外,分离抗体可实质上不含其它细胞材料和/或化学品。
“单克隆抗体”或“单克隆抗体组合物”意指对特定表位显示单一结合特异性和亲和力的单一分子组成的抗体分子制剂。
“人抗体”意指具有框架区与CDR区二者(和恒定区,如果存在)都源于人种系免疫球蛋白序列的可变区的抗体。人抗体可包括后期修饰,包括天然或合成修饰。人抗体可包括不由人种系免疫球蛋白序列编码的氨基酸残基(例如通过体外随机诱变或位点特异性诱变或通过体内体细胞突变引入的突变)。然而,“人抗体”不包括源于另一哺乳动物物种(例如小鼠)种系的CDR序列已移植至人框架序列上的抗体。
“人单克隆抗体”意指具有框架区与CDR区二者都源于人种系免疫球蛋白序列的可变区的显示单一结合特异性的抗体。在一个实施方案中,人单克隆抗体通过包括与永生细胞融合的从转基因非人动物(例如转基因小鼠)获得的B细胞的杂交瘤产生,该转基因非人动物具有包含人重链转基因和轻链转基因的基因组。
“脂(肪)族”意指具有指定碳原子数(例如在“C3脂族”、“C1-C5脂族”或“C1至C5脂族”中,后两个短语对于具有1至5个碳原子的脂族部分而言同义)或当未明确指定碳原子数时,具有1至4个碳原子(在不饱和脂族部分的情况下,2至4个碳)的直链或支链、饱和或不饱和非芳族烃部分。
“烷基”意指饱和脂族部分,且指定碳原子数的相同惯例是适用的。通过说明的方式,C1-C4烷基部分包括但不限于甲基、乙基、丙基、异丙基、异丁基、叔丁基、1-丁基、2-丁基等。“亚烷基”意指烷基的二价对应物,例如CH2CH2、CH2CH2CH2和CH2CH2CH2CH2。
“烯基”意指具有至少一个碳-碳双键的脂族部分,且指定碳原子数的相同惯例是适用的。通过说明的方式,C2-C4烯基部分包含但不限于乙烯基(ethenyl、vinyl)、2-丙烯基(烯丙基或丙-2-烯基)、顺式-1-丙烯基、反式-1-丙烯基、E- (或Z-) 2-丁烯基、3-丁烯基、1,3-丁二烯基(丁-1,3-二烯基)等。
“炔基”意指具有至少一个碳-碳三键的脂族部分,且指定碳原子数的相同惯例是适用的。通过说明的方式,C2-C4炔基包括乙炔基(ethynyl、acetylenyl)、炔丙基(丙-2-炔基)、1-丙炔基、丁-2-炔基等。
“环脂族”意指具有1至3个环的饱和或不饱和非芳族烃部分,每一环具有3至8 (优选地3至6个)碳原子。“环烷基”意指每一环都饱和的环脂族部分。“环烯基”意指至少一个环具有至少一个碳-碳双键的环脂族部分。“环炔基”意指至少一个环具有至少一个碳-碳三键的环脂族部分。通过说明的方式,环脂族部分包括但不限于环丙基、环丁基、环戊基、环戊烯基、环己基、环己烯基、环庚基、环辛基和金刚烷基。优选环脂族部分是环烷基,尤其环丙基、环丁基、环戊基和环己基。“亚环烷基”意指环烷基的二价对应物。
“杂环脂族”意指在至少一个环中至多3 (优选地1至2)个碳被独立地选自N、O或S的杂原子代替的环脂族部分,其中N和S任选地可被氧化且N任选地可被季铵化。类似地,“杂环烷基”、“杂环烯基”和“杂环炔基”分别意指至少一个环已经被如此修饰的环烷基、环烯基或环炔基部分。示例性杂环脂族部分包括氮杂环丙基、氮杂环丁基、1,3-二氧杂环己烷基、氧杂环丁基、四氢呋喃基、吡咯烷基、哌啶基、哌嗪基、四氢吡喃基、四氢硫代吡喃基、四氢噻喃基砜、吗啉基、硫代吗啉基、硫代吗啉基亚砜、硫代吗啉基砜、1,3-二氧杂环戊烷基、四氢-1,1-二氧代噻吩基、1,4-二氧杂环己烷基、硫杂环丁基(thietanyl)等。“亚杂环烷基”意指杂环烷基的二价对应物。
“烷氧基”、“芳氧基”、“烷硫基”和“芳硫基”分别意指-O(烷基)、-O(芳基)、-S(烷基)和-S(芳基)。实例分别是甲氧基、苯氧基、甲硫基和苯硫基。
“卤素”或“卤代”意指氟、氯、溴或碘。
“芳基”意指如下具有单环、双环或三环系统的烃部分:其中每一环具有3至7个碳原子且至少一个环是芳族的。环系统中的环可彼此稠合(如在萘基中)或彼此键合(如在联苯基中)且可稠合或键合至非芳族环(如在二氢茚基或环己基苯基中。通过进一步说明的方式,芳基部分包括但不限于苯基、萘基、四氢萘基、二氢茚基、联苯基、菲基、蒽基和苊基。“亚芳基”意指芳基的二价对应物,例如1,2-亚苯基、1,3-亚苯基或1,4-亚苯基。
“杂芳基”意指具有单环、双环或三环系统的如下部分:其中每一环具有3至7个碳原子且至少一个环是含有1至4个独立地选自N、O或S的杂原子的芳族环,其中N和S任选地可被氧化且N任选地可被季铵化。此类含有至少一个杂原子的芳族环可稠合至其它类型环(如在苯并呋喃基或四氢异喹啉基中)或直接键合至其它类型环(如在苯基吡啶基或2-环戊基吡啶基中)。通过进一步说明的方式,杂芳基部分包括吡咯基、呋喃基、噻吩基(thiophenyl、thienyl)、咪唑基、吡唑基、噁唑基、异噁唑基、噻唑基、异噻唑基、三唑基、四唑基、吡啶基、N-氧代吡啶基、哒嗪基、嘧啶基、吡嗪基、喹啉基、异喹啉基、喹唑啉基、噌啉基、喹喔啉基、萘啶基、苯并呋喃基、吲哚基、苯并噻吩基、噁二唑基、噻二唑基、吩噻唑基、苯并咪唑基、苯并三唑基、二苯并呋喃基、咔唑基、二苯并噻吩基、吖啶基等。“亚杂芳基”意指芳基的二价对应物。
当指示部分可取代(例如通过使用“未取代或取代”或“任选地取代”措辞,如在“未取代或取代的C1-C5烷基”或“任选地取代的杂芳基”中)时,此类部分可具有一个或多个独立选择的取代基、优选地一个至五个的数量、更优选地一个或两个的数量。本领域普通技术人员可考虑连接取代基的部分来选择取代基和取代模式以提供在化学上稳定且可通过本领域已知技术以及本文所记载方法合成的化合物。
“芳基烷基”、“(杂环脂族)烷基”、“芳基烯基”、“芳基炔基”、“联芳基烷基”等意指根据情况被芳基、杂环脂族基团、联芳基等部分取代的烷基、烯基或炔基部分,其中根据情况在烷基、烯基或炔基部分处具有开放(不饱和)化合价,例如如苄基、苯乙基、N-咪唑基乙基、N-吗啉基乙基等中。相反,“烷基芳基”、“烯基环烷基”等意指根据情况被烷基、烯基等部分取代的芳基、环烷基等部分,其根据情况为例如如甲基苯基(甲苯基)或烯丙基环己基中。“羟基烷基”、“卤代烷基”、“烷基芳基”、“氰基芳基”等意指根据情况被一个或多个指定取代基(根据情况为羟基、卤代等)取代的烷基、芳基等部分。
例如,可允许的取代基包括但不限于烷基(尤其甲基或乙基)、烯基(尤其烯丙基)、炔基、芳基、杂芳基、环脂族基团、杂环脂族基团、卤代(尤其氟)、卤代烷基(尤其三氟甲基)、羟基、羟基烷基(尤其羟乙基)、氰基、硝基、烷氧基、-O(羟基烷基)、-O(卤代烷基)(尤其-OCF3)、-O(环烷基)、-O(杂环烷基)、-O(芳基)、烷硫基、芳硫基、=O、=NH、=N(烷基)、=NOH、=NO(烷基)、-C(=O)(烷基)、-C(=O)H、-CO2H、-C(=O)NHOH、-C(=O)O(烷基)、-C(=O)O(羟基烷基)、-C(=O)NH2、-C(=O)NH(烷基)、-C(=O)N(烷基)2、-OC(=O)(烷基)、-OC(=O)(羟基烷基)、-OC(=O)O(烷基)、-OC(=O)O(羟基烷基)、-OC(=O)NH2、-OC(=O)NH(烷基)、-OC(=O)N(烷基)2、叠氮基、-NH2、-NH(烷基)、-N(烷基)2、-NH(芳基)、-NH(羟基烷基)、-NHC(=O)(烷基)、-NHC(=O)H、-NHC(=O)NH2、-NHC(=O)NH(烷基)、-NHC(=O)N(烷基)2、-NHC(=NH)NH2、-OSO2(烷基)、-SH、-S(烷基)、-S(芳基)、-S(环烷基)、-S(=O)烷基、-SO2(烷基)、-SO2NH2、-SO2NH(烷基)、-SO2N(烷基)2等。
当所取代部分是脂族部分时,优选取代基是芳基、杂芳基、环脂族基团、杂环脂族基团、卤代、羟基、氰基、硝基、烷氧基、-O(羟基烷基)、-O(卤代烷基)、-O(环烷基)、-O(杂环烷基)、-O(芳基)、烷硫基、芳硫基、=O、=NH、=N(烷基)、=NOH、=NO(烷基)、-CO2H、-C(=O)NHOH、-C(=O)O(烷基)、-C(=O)O(羟基烷基)、-C(=O)NH2、-C(=O)NH(烷基)、-C(=O)N(烷基)2、-OC(=O)(烷基)、-OC(=O)(羟基烷基)、-OC(=O)O(烷基)、-OC(=O)O(羟基烷基)、-OC(=O)NH2、-OC(=O)NH(烷基)、-OC(=O)N(烷基)2、叠氮基、-NH2、-NH(烷基)、-N(烷基)2、-NH(芳基)、-NH(羟基烷基)、-NHC(=O)(烷基)、-NHC(=O)H、-NHC(=O)NH2、-NHC(=O)NH(烷基)、-NHC(=O)N(烷基)2、-NHC(=NH)NH2、-OSO2(烷基)、-SH、-S(烷基)、-S(芳基)、-S(=O)烷基、-S(环烷基)、-SO2(烷基)、-SO2NH2、-SO2NH(烷基)和-SO2N(烷基)2。更优选取代基是卤代、羟基、氰基、硝基、烷氧基、-O(芳基)、=O、=NOH、=NO(烷基)、-OC(=O)(烷基)、-OC(=O)O(烷基)、-OC(=O)NH2、-OC(=O)NH(烷基)、-OC(=O)N(烷基)2、叠氮基、-NH2、-NH(烷基)、-N(烷基)2、-NH(芳基)、-NHC(=O)(烷基)、-NHC(=O)H、-NHC(=O)NH2、-NHC(=O)NH(烷基)、-NHC(=O)N(烷基)2和-NHC(=NH)NH2。特别优选的是苯基、氰基、卤代、羟基、硝基、C1-C4烷氧基、O(C2-C4亚烷基)OH和O(C2-C4亚烷基)卤代。
当所取代部分是环脂族基团、杂环脂族基团、芳基或杂芳基部分时,优选取代基是烷基、烯基、炔基、卤代、卤代烷基、羟基、羟基烷基、氰基、硝基、烷氧基、-O(羟基烷基)、-O(卤代烷基)、-O(芳基)、-O(环烷基)、-O(杂环烷基)、烷硫基、芳硫基、-C(=O)(烷基)、-C(=O)H、-CO2H、-C(=O)NHOH、-C(=O)O(烷基)、-C(=O)O(羟基烷基)、-C(=O)NH2、-C(=O)NH(烷基)、-C(=O)N(烷基)2、-OC(=O)(烷基)、-OC(=O)(羟基烷基)、-OC(=O)O(烷基)、-OC(=O)O(羟基烷基)、-OC(=O)NH2、-OC(=O)NH(烷基)、-OC(=O)N(烷基)2、叠氮基、-NH2、-NH(烷基)、-N(烷基)2、-NH(芳基)、-NH(羟基烷基)、-NHC(=O)(烷基)、-NHC(=O)H、-NHC(=O)NH2、-NHC(=O)NH(烷基)、-NHC(=O)N(烷基)2、-NHC(=NH)NH2、-OSO2(烷基)、-SH、-S(烷基)、-S(芳基)、-S(环烷基)、-S(=O)烷基、-SO2(烷基)、-SO2NH2、-SO2NH(烷基)和-SO2N(烷基)2。更优选取代基是烷基、烯基、卤代、卤代烷基、羟基、羟基烷基、氰基、硝基、烷氧基、-O(羟基烷基)、-C(=O)(烷基)、-C(=O)H、-CO2H、-C(=O)NHOH、-C(=O)O(烷基)、-C(=O)O(羟基烷基)、-C(=O)NH2、-C(=O)NH(烷基)、-C(=O)N(烷基)2、-OC(=O)(烷基)、-OC(=O)(羟基烷基)、-OC(=O)O(烷基)、-OC(=O)O(羟基烷基)、-OC(=O)NH2、-OC(=O)NH(烷基)、-OC(=O)N(烷基)2、-NH2、-NH(烷基)、-N(烷基)2、-NH(芳基)、-NHC(=O)(烷基)、-NHC(=O)H、-NHC(=O)NH2、-NHC(=O)NH(烷基)、-NHC(=O)N(烷基)2和-NHC(=NH)NH2。特别优选的是C1-C4烷基、氰基、硝基、卤代和C1-C4烷氧基。
当陈述范围(如在“C1-C5烷基”或“5%至10%”中)时,此类范围包括范围的端点,如第一情况中的C1和C5和第二情况中的5%和10%。
除非明确指示特定立体异构体(例如通过结构式中相关立体中心处的粗体或虚线键,通过在结构式中将双键描绘为具有E或Z构型,或通过使用立体化学指定命名),否则所有立体异构体都作为纯化合物以及其混合物包括在本发明范围内。除非另外指示,否则本发明涵盖所有其个别对映异构体、非对映异构体、几何异构体及其组合和混合物。
本领域技术人员应理解,化合物可具有等效于本文所用结构式中描绘的那些的互变异构体形式(例如酮形式和烯醇形式)、共振形式和两性离子形式,且所述结构式涵盖此类互变异构体形式、共振形式或两性离子形式。
“药学上可接受的酯”意指在体内水解(例如在人体中)以产生母体化合物或其盐或本身具有类似于母体化合物的活性的酯。合适酯包括C1-C5烷基、C2-C5烯基或C2-C5炔基酯,尤其甲基、乙基或正丙基酯。
“药学上可接受的盐”意指适于药物制剂的化合物的盐。当化合物具有一个或多个碱性基团时,该盐可以是酸加成盐,例如硫酸盐、氢溴酸盐、酒石酸盐、甲磺酸盐、马来酸盐、柠檬酸盐、磷酸盐、乙酸盐、双羟萘酸盐(恩波酸盐(embonate))、氢碘酸盐、硝酸盐、盐酸盐、乳酸盐、甲基硫酸盐、富马酸盐、苯甲酸盐、琥珀酸盐、甲磺酸盐、乳糖酸盐、辛二酸盐、甲苯磺酸盐等。当化合物具有一个或多个酸性基团时,该盐可以是例如以下的盐:钙盐、钾盐、镁盐、葡甲胺盐、铵盐、锌盐、哌嗪盐、氨丁三醇盐、锂盐、胆碱盐、二乙胺盐、4-苯基环己胺盐、苄星盐(benzathine salt)、钠盐、四甲基铵盐等。多晶形结晶形式和溶剂化物也涵盖于本发明范围内。
组合物
在式(I) (下文出于便利性重复)中,
基团R1优选是Me、Et、n-Pr、i-Pr或
,更优选是后者。
此外在式(I)中,基团R2优选是C1-C5烷基、C1-C5烯基、C1-C5炔基、CH2OC(=O)C1-C5烷基、CH2OC(=O)C1-C5烯基、CH2OC(=O)C1-C5炔基、
或。
此外在式(I)中,优选基团N(R3a)(R3b)是:
、、、、,
、、和,
其中特别优选地,R3a和R3b中的一个是H且另一个是Me。在其它优选实施方案中,R3a和R3b都是H或都是Me,或R3a和R3b中的一个是H且另一个是C6H5。
在另一优选实施方案中,R3a和R3b独立地是H、C1-C5烷基、CH2(C5-C6环烷基)、CH2C6H5或CH2CH2OH。
在式(I)中的R1和R2的定义中,当将基团定义为未取代或取代时,其优选未取代。
在本说明书的式中,穿过苯基环在苯基环的两个碳之间的键意指连接至键的基团可位于苯基环的邻位、间位或对位中的任一处。通过说明的方式,式
代表
、或。
具有各种R2和R4基团的妥布赖森Tuv和Tup亚单元的对应物的合成由Cheng等人,2011教导,其公开内容通过引用并入本文。
在根据式(I)的化合物的优选实施方案中,R1是
,
R2是C1-C5烷基(尤其Me或n-Pr)或
;
R3a和R3b中的一个是H且另一个是Me;R4是
,
其中Y是H或NO2且R4a是H、Me或Et;
R5是Me;W是O,且n是1。
在根据式(I)的化合物的另一优选实施方案中,n是1,W是O,R4中的Y是H或NO2 (优选地H),且R2是
和更优选地。
根据该优选实施方案的化合物由式(Ia)代表:
,
其中Y是H或NO2;R4a是H、Me或Et;且R3a和R3b独立地是H、C6H5、Me或Et;或其药学上可接受的盐。
甚至更优选地,化合物具有由式(Ia')代表的结构:
,
其中R4a是H、Me或Et;且R3a和R3b独立地是H、C6H5、Me或Et;或其药学上可接受的盐。
在又一优选实施方案中,W是O,Y是NH2,n是1,且R2中的两个基团R2a并非NH2。根据该实施方案的化合物由式(Ib)代表:
,
其中R4a是H、Me或Et;R3a和R3b独立地是H、C6H5、Me和Et;且R6是C1-C5烷基、CH2OC(=O)C1-C5烷基或(CH2)1-2C6H5;或其药学上可接受的盐。
在又一优选实施方案中,化合物具有由式(Ib')代表的结构:
,
其中R4a是H、Me或Et (优选地H)且R6是Me或n-Pr;或其药学上可接受的盐。
优选地,在式(Ia)、(Ia')和(Ib)中,R3a和R3b中的一个是H且另一个是Me。在其它优选实施方案中,R3a和R3b都是H或都是Me,或R3a和R3b中的一个是H且另一个是C6H5。在其它优选实施方案中,R3a和R3b独立地是H、Me或Et。
本发明化合物的具体实例包含紧接下文显示的化合物连同其药学上可接受的盐:
、
、
、
、
、
、
、
、
、
、
和
。
化合物(I-2)、(I-7)、(I-8)和(I-9)是优选的。
缀合物
任选地,本发明化合物可缀合至特异性或优先结合至癌细胞上的化学实体的靶向部分。优选地,靶向部分是抗体或其抗原结合部分且化学实体是肿瘤相关性抗原。优选地,经由至Tuv或Tup亚单元中的官能团(例如氨基)的化学键来实现缀合。
在另一个实施方案中,提供由式(II)代表的缀合物,其包含根据本发明的细胞毒性化合物和配体,
[D(XD)aC(XZ)b]mZ (II)
其中Z是配体;D是根据本发明的细胞毒性化合物(例如根据式(I)、(Ia)、(Ia')或(Ib)的化合物);且-(XD)aC(XZ)b-通称为“接头部分”或“接头”,因为其连接Z和D。在接头内,C是经设计在化合物D的预期生物作用位点处或附近裂解的可裂解基团;XD和XZ称为间隔基部分(或“间隔基”),因为其分别使D与C和C与Z间隔开;下标a和b独立地是0或1 (即,XD和/或XZ的存在是任选的);且下标m是1、2、3、4、5、6、7、8、9或10 (优选地1、2、3或4)。D、XD、C、XZ和Z更全面地描述于下文中。
配体Z (例如抗体)发挥靶向功能。通过结合至抗原或受体所定位的靶组织或细胞,配体Z将缀合物引向那里。优选地,靶组织或细胞是癌症组织或细胞且抗原或受体是肿瘤相关性抗原,即与非癌细胞相比由癌细胞独特表达或由癌细胞过表达的抗原。基团C在靶组织或细胞处的裂解释放化合物D以在局部施加其细胞毒性效应。在一些情况下,缀合物通过胞吞作用内在化至靶细胞中且在靶细胞内发生裂解。以该方式,在预期作用位点处实现化合物D的精确递送,从而减小所需剂量。此外,化合物D在其缀合状态时通常是无生物活性的(或活性显著较小),由此减小对于非靶组织或细胞的不期望毒性。由于抗癌药通常对细胞具有高毒性,所以这是重要考虑。
如通过下标m所反映,根据配体Z具有可用于偶联的位点数量和所采用的实验条件,每一配体Z分子可与多于一个化合物D分子缀合。本领域技术人员应理解,尽管每一个别配体Z分子缀合至整数数量的化合物D,但可分析缀合物制剂的化合物D对配体Z的非整数比率,其反映了统计学平均值。该比率称为替换比(SR)或另外在抗体-药物缀合物的情况下称为药物-抗体比(DAR)。
配体Z和其缀合
优选地,配体Z是抗体。出于便利性和简单性且并不加以限制,本文随后关于配体Z偶联的详细论述是在其是抗体的背景下来书写,但本领域技术人员应理解,可偶联其它类型的配体Z (加以必要的变通)。例如,具有叶酸作为配体的缀合物可靶向在表面上具有叶酸盐受体的细胞(Vlahov等人,2008;Leamon等人,2008)。出于相同原因,下文的详细论述主要以1:1比率的抗体Z与化合物D (m = 1)的形式来书写。
优选地,配体Z是针对肿瘤相关性抗原的抗体,允许包含此类配体Z的缀合物选择性靶向癌细胞。此类抗原的实例包含:间皮素(mesothelin)、前列腺特异性膜抗原(PSMA)、CD19、CD22、CD30、CD70、B7H4 (也称为O8E)、蛋白酪氨酸激酶7 (PTK7)、磷脂酰肌醇蛋白聚糖-3 (glypican-3)、RG1、CTLA-4和CD44。抗体可以是动物(例如鼠)抗体、嵌合抗体、人源化抗体或优选地人抗体。抗体优选是单克隆抗体、尤其单克隆人抗体。针对一些上述抗原的人单克隆抗体的制备公开于:Korman等人,US 2009/0074660 A1 (B7H4);Rao-Naik等人,8,097,703 B2 (CD19);King等人,US 2010/0143368 A1 (CD22);Keler等人,US 7,387,776 B2 (2008) (CD30);Terrett等人,US 8,124,738 B2 (CD70);Korman等人,US 6,984,720 B1 (2006) (CTLA-4);Korman等人,US 8,008,449 B2 (2011) (PD-1);Huang等人,US 2009/0297438 A1和Cardarelli等人,US 7,875,278 B2 (PSMA);Terrett等人,US 2010/0034826 A1 (PTK7);Terrett等人,US 2010/0209432 (A1) (磷脂酰肌醇蛋白聚糖-3);Harkins等人,US 7,335,748 B2(2008) (RG1);Terrett等人,US 8,268,970 B2 (2012) (间皮素);和Xu等人,US 2010/0092484 A1 (CD44);其公开内容通过引用并入本文。
配体Z也可以是抗体片段或抗体模拟物,例如亲和体、结构域抗体(dAb)、纳米抗体、单抗体(unibody)、DARPin、抗运载蛋白(anticalin)、万能抗体(versabody)、多卡林(duocalin)、脂质运载蛋白(lipocalin)或阿维莫(avimer)。
配体Z上的几种不同反应性基团中的任一种都可以是缀合位点,包括赖氨酸残基中的ε-氨基、侧链碳水化合物部分、羧酸基团、二硫化物基团和硫醇基团。每一类型的反应性基团代表具有一些优点和一些缺点的折衷。关于适于缀合的抗体反应性基团的综述,参见例如Garnett, Adv. Drug Delivery Rev. 53 (2001), 171-216和Dubowchik与Walker,Pharmacology & Therapeutics 83 (1999), 67-123,其公开内容通过引用并入本文。
在一个实施方案中,经由赖氨酸ε-氨基使配体Z缀合。大部分抗体具有多个暴露的赖氨酸ε-氨基,其可经由酰胺、脲、硫脲或氨基甲酸酯键使用本领域已知的技术(包括使用异双功能试剂进行修饰)缀合(如下文中进一步描述)。然而,难以控制何种和多少ε-氨基发生反应,从而导致在缀合物制备中具有潜在的批次间变化性。此外,缀合可导致对于维持抗体原始构型重要的质子化ε-氨基的中和或可发生于抗原结合位点附近或抗原结合位点处的赖氨酸,二者都不是期望事件。
在另一个实施方案中,配体Z可经由碳水化合物侧链缀合,因为许多抗体是糖基化的。碳水化合物侧链可用高碘酸盐氧化以生成醛基团,该醛基团继而可与胺反应形成亚胺基团,例如在缩氨基脲、肟或腙中。如果期望,则可通过使用氰基硼氢化钠还原来将亚胺基团转化成更稳定的胺基团。关于经由碳水化合物侧链缀合的其它公开内容,参见例如Rodwell等人,Proc. Nat'l Acad. Sci. USA 83, 2632-2636 (1986);其公开内容通过引用并入本文。如同对于赖氨酸ε-氨基而言,也存在关于缀合位点位置的重现性和化学计量学的问题。
在又一个实施方案中,配体Z可经由羧酸基团缀合。在一个实施方案中,对末端羧酸基团官能化以生成碳酰肼,然后使碳酰肼与具有醛的缀合部分反应。参见Fisch等人,Bioconjugate Chemistry 1992, 3, 147-153。
在又一个实施方案中,可经由使抗体Z上的半胱氨酸残基和缀合物另一部分上的硫进行二硫化物基团桥接来使抗体Z缀合。一些抗体缺乏游离硫醇(巯基)基团但具有二硫化物基团,例如在铰链区中。在此类情况下,可通过还原原始二硫化物基团来生成游离硫醇基团。如此生成的硫醇基团然后可用于缀合。参见例如Packard等人,Biochemistry 1986, 25, 3548-3552;King等人,Cancer Res. 54, 6176-6185 (1994);和Doronina等人,Nature Biotechnol. 21(7), 778-784 (2003);其公开内容通过引用并入本文。同样,存在关于缀合位点位置和化学计量学以及可能破坏抗体原始构型的问题。
已知将游离硫醇基团引入抗体中而不断裂原始二硫化物键的许多方法,所述方法可使用本发明的配体Z进行实践。取决于所采用方法,可在预定位置处引入可预计数量的游离巯基。在一种方法中,制备用半胱氨酸代替另一氨基酸的突变抗体。参见例如Eigenbrot等人,US 7,521,541 B2 (2009);Chilkoti等人,Bioconjugate Chem. 1994, 5, 504-507;Urnovitz等人,US 4,698,420 (1987);Stimmel等人,J. Biol. Chem., 275 (39), 30445-30450 (2000);Bam等人,US 7,311,902 B2 (2007);Kuan等人,J. Biol. Chem., 269 (10), 7610-7618 (1994);Poon等人,J. Biol. Chem., 270 (15), 8571-8577 (1995)。在另一方法中,将额外半胱氨酸添加至C-末端。参见例如Cumber等人,J. Immunol., 149, 120-126 (1992);King等人,Cancer Res., 54, 6176-6185 (1994);Li等人,Bioconjugate Chem., 13, 985-995 (2002);Yang等人, Protein Engineering, 16, 761-770 (2003);和Olafson等人,Protein Engineering Design & Selection, 17, 21-27 (2004)。Liu等人于WO 2009/026274 A1中教导了引入游离半胱氨酸的优选方法,其中将具有半胱氨酸的氨基酸序列添加至抗体重链的C-末端。该方法在远离抗原结合位点的已知位置处引入已知数量的半胱氨酸残基(每条重链一个残基)。本段落中引用文件的公开内容都通过引用并入本文。
在又一个实施方案中,可使用异双功能试剂(例如2-亚氨基硫杂环戊烷或N-琥珀酰亚胺基-3-(2-吡啶基二硫代)丙酸酯(SPDP))修饰赖氨酸ε-氨基,从而将ε-氨基转化成硫醇或二硫化物基团,如原样产生半胱氨酸替代物。然而,该方法具有与ε-氨基本身(proper)有关的相同缀合位置和化学计量学限制的问题。
在又一优选实施方案中,经由硫醇基团至受体部分的亲核性加成产物使配体Z缀合。优选受体部分是马来酰亚胺基团,其与抗体硫醇基团的反应一般说明于下文中。硫醇基团可以是原始基团或如上文所述引入的基团。
还可经由适用于“点击”化学的官能团使配体Z缀合,如下文所讨论。
接头–(XD)aC(XZ)b–
如上所述,本发明缀合物的接头部分包含至多三种要素:可裂解基团C和任选间隔基XZ和XD。
可裂解基团C是可在生理学条件下裂解的基团,优选地选择其以便在缀合物通常在血浆中循环的同时其相对稳定,而一旦缀合物到达其预期作用部位(即靠近靶细胞、在靶细胞处或靶细胞内),其易于裂解。优选地,在使抗体Z结合至展示于靶细胞表面的抗原上后,通过胞吞作用使缀合物由靶细胞内在化。随后,基团C在靶细胞的泡囊体(早期核内体、晚期核内体或尤其溶酶体)发生裂解。
在一个实施方案中,基团C是pH敏感性基团。血浆中的pH略高于中性,而溶酶体内侧的pH是酸性(约5)。因此,酸催化裂解的基团C在溶酶体内侧的裂解速率比血浆速率快几个数量级。合适酸敏感性基团的实例包含顺式-乌头酰胺和腙,如描述于:Shen等人,US 4,631,190 (1986);Shen等人,US 5,144,011 (1992);Shen等人,Biochem. Biophys. Res. Commun. 102, 1048-1054 (1981)和Yang等人,Proc. Natl Acad. Sci (USA), 85, 1189-1193 (1988);其公开内容通过引用并入本文。
在另一个实施方案中,基团C是二硫化物。二硫化物可通过硫醇-二硫化物交换机制以取决于环境硫醇浓度的速率裂解。由于谷胱甘肽和其它硫醇的细胞内浓度高于其血清浓度,所以二硫化物在细胞内的裂解速率较高。此外,可通过调整二硫化物的立体和电子特征(例如烷基-芳基二硫化物相比于烷基-烷基二硫化物;在芳基环上取代等)来调节硫醇-二硫化物交速换率,从而使得能够设计具有增强的血清稳定性或特定裂解速率的二硫键。关于涉及缀合物中的二硫化物可裂解基团的其它公开内容,参见例如Thorpe等人,Cancer Res. 48, 6396-6403 (1988);Santi等人,US 7,541,530 B2 (2009);Ng等人,US 6,989,452 B2 (2006);Ng等人,WO 2002/096910 A1;Boyd等人,US 7,691,962 B2;和Sufi等人,US 2010/0145036 A1;其公开内容通过引用并入本文。
优选基团C包含与在血清中由蛋白酶裂解相比优先在预期作用位点处由蛋白酶裂解的肽键。通常,基团C包含1至20个氨基酸、优选地1至6个氨基酸、更优选地1至3个氨基酸。氨基酸可以是天然和/或非天然α-氨基酸。天然氨基酸是由基因密码编码的那些以及由其衍生的氨基酸,例如羟基脯氨酸、γ-羧基谷氨酸盐、瓜氨酸和O-磷酸丝氨酸。术语氨基酸还包括氨基酸类似物和模拟物。类似物是与天然氨基酸具有相同一般H2N(R)CHCO2H结构的化合物,除了R基团不是天然氨基酸中发现的基团。类似物的实例包括高丝氨酸、正亮氨酸、甲硫氨酸亚砜和甲硫氨酸甲基锍。氨基酸模拟物是具有不同于α-氨基酸的一般化学结构的结构但以与其类似方式发挥作用的化合物。术语“非天然氨基酸”意欲代表“D”立体化学形式,天然氨基酸是“L”形式。
优选地,基团C含有作为用于蛋白酶的裂解识别序列的氨基酸序列。本领域已知许多裂解识别序列。参见例如Matayoshi等人,Science 247: 954 (1990);Dunn等人,Meth. Enzymol. 241: 254 (1994);Seidah等人,Meth. Enzymol. 244: 175 (1994);Thornberry, Meth. Enzymol. 244: 615 (1994);Weber等人,Meth. Enzymol. 244: 595 (1994);Smith等人,Meth. Enzymol. 244: 412 (1994);和Bouvier等人,Meth. Enzymol.248: 614 (1995);其公开内容通过引用并入本文。
对于并不意欲由细胞内在化的缀合物而言,可选择基团C以便其由靶组织附近的细胞外基质中存在的蛋白酶(例如由附近死细胞释放的蛋白酶或肿瘤相关蛋白酶)裂解。示例性细胞外肿瘤相关蛋白酶是基质金属蛋白酶(MMP)、甲拌磷寡肽酶(TOP)和CD10。
对于经设计由细胞内在化的缀合物而言,基团C优选地包含经选择用于由内体或溶酶体蛋白酶(尤其后者)裂解的氨基酸序列。此类蛋白酶的非限制性实例包括组织蛋白酶B、C、D、H、L和S,尤其组织蛋白酶B。组织蛋白酶B优先在序列-AA2-AA1-处裂解肽,其中AA1是碱性或强氢键键合氨基酸(例如赖氨酸、精氨酸或瓜氨酸)且AA2是疏水性氨基酸(例如苯丙氨酸、缬氨酸、丙氨酸、亮氨酸或异亮氨酸),例如Val-Cit (其中Cit表示瓜氨酸)或Val-Lys。(在本文中,除非上下文另外明确指示,否则氨基酸序列是以N-至-C方向书写,如在H2N-AA2-AA1-CO2H中。)关于组织蛋白酶可裂解基团的其它信息,参见Dubowchik等人,Biorg. Med. Chem. Lett. 8, 3341-3346 (1998);Dubowchik等人,Bioorg. Med. Chem. Lett., 8 3347-3352 (1998);和Dubowchik等人,Bioconjugate Chem. 13, 855-869 (2002);其公开内容通过引用并入本文。另一可用于裂解肽基接头的酶是豆球蛋白(legumain),其是优先在Ala-Ala-Asn处裂解的溶酶体半胱氨酸蛋白酶。
在一个实施方案中,基团C是包含二氨基酸序列-AA2-AA1-的肽,其中AA1是赖氨酸、精氨酸或瓜氨酸且AA2是苯丙氨酸、缬氨酸、丙氨酸、亮氨酸或异亮氨酸。在另一个实施方案中,C由一个至五个氨基酸的序列组成,其选自:Val-Cit、Ala-Val、Val-Ala-Val、Lys-Lys、Ala-Asn-Val、Val-Leu-Lys、Cit-Cit、Val-Lys、Ala-Ala-Asn、Lys、Cit、Ser和Glu。
由单一氨基酸组成的可裂解基团C的制备和设计公开于Chen等人,US 2010/0113476 A1中,其公开内容通过引用并入本文。
基团C也可以是在暴露于光后裂解的光可裂解基团,例如硝基苄基醚。
基团C可直接键合至抗体Z或化合物D;即,间隔基XZ和XD根据情况可不存在。例如,如果基团C是二硫化物,则两个硫中的一个可以是抗体Z上的半胱氨酸残基或其替代物。或者,基团C可以是键合至抗体的碳水化合物侧链上的醛的腙。或者,基团C可以是使用抗体Z的赖氨酸ε-氨基形成的肽键。在优选实施方案中,化合物D经由至化合物D中的羧基或胺基团的肽基键直接键合至基团C。
当存在时,间隔基XZ提供基团C与抗体Z之间的空间分离,以免前者在立体上干扰后者结合抗原或后者在立体上干扰前者的裂解。此外,可使用间隔基XZ来赋予缀合物增加的溶解度或降低的聚集性质。间隔基XZ可包含一个或多个可以任何数量的组合装配的模块区段。适用于间隔基XZ的区段的实例是:
、、、
、、
和其组合,
其中下标q是0或1且下标r是1至24、优选地2至4。可组合这些区段,例如下文所显示:
、
或。
间隔基XD,如果存在,提供基团C和化合物D之间的空间分离,以免后者在立体上或在电子上干扰前者的裂解。间隔基XD也可用于向缀合物中引入额外分子量和化学功能性。通常,额外质量和功能性将影响缀合物的血清半衰期和其它性质。因此,经由明智选择间隔基基团,可调节缀合物的血清半衰期。间隔基D也可从模块区段装配,如上文在间隔基XZ的背景下所述。
间隔基XZ和/或XD,如果存在,优选地分别在Z和C或D和C之间提供4至25个原子、更优选4至20个原子的线性分离。
间隔基XZ或XD或二者可包括自我牺牲部分(self-immolating moiety)。自我牺牲部分是如下部分:(1)其键合至基团C和抗体Z或细胞毒素D,且(2)其结构使得从基团C的裂解引发导致自我牺牲部分本身根据情况从抗体Z或细胞毒素D去键合(disbonding)的反应序列。换言之,在远离抗体Z或细胞毒素D的位点处的反应(从基团C裂解)也导致XZ-Z或XD-D键破裂。自我牺牲部分的存在在间隔基XD的情况下是期望的,因为如果在缀合物裂解之后间隔基XD或其部分欲保持连接至细胞毒素D,则后者的生物活性可受损。当可裂解基团C是多肽时,自我牺牲部分的使用尤其是期望的。
键合至配偶体分子D上的羟基或氨基的示例性自我牺牲部分(i)-(v)显示于下文中:
。
自我牺牲部分是虚线a与b之间的结构,其中显示毗邻结构特征以提供背景。自我牺牲部分(i)和(v)键合至化合物D-NH2 (即化合物D经由氨基缀合),而自我牺牲部分(ii)、(iii)和(iv)键合至化合物D-OH (即化合物D经由羟基或羧基缀合)。虚线b处的酰胺键的裂解释放酰胺氮作为胺氮,从而引发导致虚线a处的键裂解且随后根据情况释放D-OH或D-NH2的反应序列。关于自我牺牲部分的其它公开内容,参见Carl等人,J. Med. Chem., 24 (3), 479-480 (1981);Carl等人,WO 81/01145 (1981);Dubowchik等人,Pharmacology & Therapeutics, 83, 67-123 (1999);Firestone等人,US 6,214,345 B1 (2001);Toki等人,J. Org. Chem. 67, 1866-1872 (2002);Doronina等人,Nature Biotechnology 21 (7), 778-784 (2003) (勘误,第941页);Boyd等人,US 7,691,962 B2;Boyd等人,US 2008/0279868 A1;Sufi等人,WO 2008/083312 A2;Feng, US 7,375,078 B2;和Senter等人,US 2003/0096743 A1;其公开内容通过引用并入本文。
在另一个实施方案中,抗体靶向部分和细胞毒性化合物D通过不可裂解接头连接。抗体的降解最终将接头减小为不干扰细胞毒性化合物D的生物活性的小附加部分。
化合物D -接头组合物
优选地通过首先使化合物D和接头(XD)aC(XZ)b (其中XD、C、XZ、a和b如针对式(II)所定义)连接以形成由式(III)代表的药物-接头组合物来制备本发明缀合物:
D-(XD)aC(XZ)b-R31 (III)
其中R31是适于与抗体Z上的官能团反应形成缀合物的官能团。合适基团R31的实例包括氨基、叠氮化物、环辛炔、
、、、、、、、、和;
其中R32是Cl、Br、F、甲磺酸根或甲苯磺酸根且R33是Cl、Br、I、F、OH、-O-N-琥珀酰亚胺基、-O-(4-硝基苯基)、-O-五氟苯基或-O-四氟苯基。通常可用于制备合适部分D-(XD)aC(XZ)b-R31的化学品公开于:Ng等人,US 7,087,600 B2 (2006);Ng等人,US 6,989,452 B2 (2006);Ng等人,US 7,129,261 B2 (2006);Ng等人,WO 02/096910 A1;Boyd等人,US 7,691,962 B2;Chen等人,US 7,517,903 B2 (2009);Gangwar等人,US 7,714,016 B2 (2010);Boyd等人,US 2008/0279868 A1;Gangwar等人,US 7,847,105 B2 (2010);Gangwar等人,US 7,968,586 B2 (2011);Sufi等人,US 2010/0145036 A1;和Chen等人,US 2010/0113476 A1;其公开内容通过引用并入本文。
优选地,反应性官能团-R31是-NH2、-OH、-CO2H、-SH、马来酰亚胺基、环辛炔、叠氮基(-N3)、羟基氨基(-ONH2)或N-羟基琥珀酰亚胺基。特别优选官能团-R31选自:
。
-OH基团可被抗体上(例如,天冬氨酸或谷氨酸侧链上)的羧基酯化。
-CO2H基团可被抗体上的-OH基团酯化或被抗体上(例如赖氨酸侧链上)的氨基酰胺化。
N-羟基琥珀酰亚胺基团是在功能上被活化的羧基且可通过与氨基(例如来自赖氨酸)的反应便利地酰胺化。
马来酰亚胺基团可在Michael加成反应中与抗体上的-SH基团(例如来自半胱氨酸或来自引入巯基官能团的抗体的化学修饰)缀合。
-SH基团当抗体已经修饰以向其中引入马来酰亚胺基团时在Michael加成反应(其是上述Michael加成反应的“镜像”)中对于缀合尤其有用。抗体可被4-(马来酰亚胺基甲基)-环己烷甲酸N-琥珀酰亚胺基酯(SMCC)或其磺化变体磺基-SMCC修饰以具有马来酰亚胺基团,这两种试剂都购自Sigma-Aldrich。
叠氮化物和环辛炔是可经由所谓的无铜“点击化学”实现缀合的互补官能团,其中叠氮化物加成至环辛炔的整个应变炔键上以形成1,2,3-三唑环。参见例如Agard等人,J. Amer. Chem. Soc.2004, 126, 15046-15047;Best, Biochemistry2009, 48, 6571-6584。叠氮化物可以是式(III)中的反应性官能团R31且环辛炔可位于抗体或其抗原结合部分上,或反之亦然。环辛炔基团可通过DIBO试剂(购自Invitrogen/Molecular Probes, Eugene, Oregon)提供。
可利用将非天然氨基酸引入抗体中的技术,其中非天然氨基酸提供用于与反应性官能团缀合的官能团。例如,可将非天然氨基酸对乙酰基苯丙氨酸并入抗体或其它多肽中,如Tian等人,WO 2008/030612 A2 (2008)中所教导。对乙酰基苯丙氨酸中的酮基团通过与羟基氨基反应性官能团形成肟可以是缀合位点。或者,可将非天然氨基酸对叠氮基苯丙氨酸并入抗体中以提供用于经由点击化学缀合的叠氮化物官能团。也可使用无细胞方法将非天然氨基酸并入抗体或其它多肽中,如Goerke等人,US 2010/0093024 A1 (2010)和Goerke等人,Biotechnol. Bioeng. 2009, 102 (2), 400-416中所教导。
胺(NH2)基团可用于使用酶转谷氨酰胺酶进行缀合,如Jeger等人,Angew. Chem. Int. Ed.2010, 49, 9995-9997中所教导。
也可使用酶转肽酶A (Sortase A)来实现缀合,如Levary等人,PLoS One2011, 6(4), e18342;Proft, Biotechnol. Lett.2010, 32, 1-10;Ploegh等人,WO 2010/087994 A2 (2010);和Mao等人,WO 2005/051976 A2 (2005)中所教导。转肽酶A识别基序(通常是LPXTG,其中X是任一天然氨基酸)可位于配体Z上且亲核性受体基序(通常是GGG)可以是式(III)中的基团R31,或反之亦然。
式[D(XD)aC(XZ)b]mZ和D-(XD)aC(XZ)b-R31中的基团D优选地具有根据以下式的结构:式(D-a)
或式(D-b)
其中Y是H或NO2;R4a是H、Me或Et;R3a和R3b独立地是H、Me或Et;且R6是C1-C5烷基、CH2OC(=O)C1-C5烷基或(CH2)1-2C6H5。
此类基团D的实例包括:
、
、和
,
其中R7是H、Me或Et。
根据式D-(XD)aC(XZ)b-R31组合物的实例包括紧接下文所显示者;连同其药学上可接受的盐:
、
、
、
、
、
、
.
、
、
、
、
、
和
。
优选药物-接头化合物具有由式(III-a)代表的结构:
其中
R3a和R3b独立地是H、Me或Et;
R6是Me、Et或n-Pr;
AAa和每一AAb独立地选自:丙氨酸、β-丙氨酸、γ-氨基丁酸、精氨酸、天冬酰胺、天冬氨酸、γ-羧基谷氨酸、瓜氨酸、半胱氨酸、谷氨酸、谷氨酰胺、甘氨酸、组氨酸、异亮氨酸、亮氨酸、赖氨酸、甲硫氨酸、正亮氨酸、正缬氨酸、鸟氨酸、苯丙氨酸、脯氨酸、丝氨酸、苏氨酸、色氨酸、酪氨酸和缬氨酸;
p是1、2、3、或4;
q是1、2、3、4、5、6、7、8、9或10 (优选地2、3或4);
r是1、2、3、4或5;
s是0或1;且
R31选自:
或其药学上可接受的盐。
在式(III-a)中,-AAa-[AAb]p-代表多肽,其长度由p值决定(例如如果p是1,则为二肽,如果p是3,则为四肽等)。AAa位于多肽的羧基末端处且其羧基与药物的苯胺基氮形成肽(酰胺)键。相反,最后AAb位于多肽的氨基末端处且其α-氨基与
(如果s是1)和
(如果s是0)形成肽键。
更优选药物-接头化合物具有由式(III-b)代表的结构:
其中
R6是Me或n-Pr;
q是1、2、3、4、5、6、7、8、9或10 (优选地2、3或4);
r是1、2、3、4或5;
s是0或1;且
R31选自:
或其药学上可接受的盐。
缀合物的制备
下文是说明性程序,其基于通过以下方式将游离硫醇基团引入抗体中:使赖氨酸ε-氨基与2-亚氨基硫杂环戊烷反应,随后与含有马来酰亚胺的药物-接头部分(例如上文所述)反应。首先将抗体以缓冲液交换至含有50 mM NaCl和2 mM二伸乙基三胺五乙酸(DTPA)的0.1 M磷酸盐缓冲液(pH 8.0)中并浓缩至5-10 mg/mL。经由将2-亚氨基硫杂环戊烷添加至抗体中来实现硫醇化。待添加2-亚氨基硫杂环戊烷的量可通过初步实验来确定且随抗体而变化。在初步实验中,将增加量的2-亚氨基硫杂环戊烷的滴定物添加至抗体中,且在RT (室温,约25℃)下与抗体孵育1 h后,使用SEPHADEX™ G-25柱将抗体脱盐至50 mM pH 6.0 HEPES缓冲液中且通过与二硫联吡啶(DTDP)反应来快速确定所引入硫醇基团的数量。硫醇基团与DTDP的反应导致硫吡啶释放,其可在324 nm下以分光光度法监测。通常使用蛋白浓度为0.5-1.0 mg/mL的样品。可使用280 nm下的吸光度来准确地确定样品中蛋白的浓度,且然后在室温下将每一样品的等分样品(0.9 mL)与0.1 mL DTDP (5 mM乙醇中的储备溶液)孵育10 min。还一同孵育单独缓冲液与DTDP的空白样品。在10 min之后,测量324 nm下的吸光度且使用19,800 M-1硫吡啶的消光系数来定量硫醇基团的数量。
通常,每一抗体约三个硫醇基团的硫醇化水平是期望的。例如,对于一些抗体而言,这可通过添加15倍摩尔过量的2-亚氨基硫杂环戊烷、随后在室温下孵育1 h来实现。然后将抗体与2-亚氨基硫杂环戊烷以期望摩尔比率孵育且然后脱盐至缀合缓冲液(含有5 mM甘氨酸和2 mM DTPA的50 mM pH 6.0 HEPES缓冲液)中。将硫醇化材料维持在冰上,同时如上所述定量所引入硫醇的数量。
在验证所引入硫醇的数量之后,以每硫醇3倍摩尔过量添加药物-接头部分。使缀合反应在还含有最终浓度为5%的二甲基亚砜(DMSO)或类似替代溶剂的缀合缓冲液中进行。通常,将药物-接头储备溶液溶于100% DMSO中。将储备溶液直接添加至具有经添加以将最终浓度达到10%的足够DMSO的硫醇化抗体中,或在含有最终浓度为10%的DMSO的缀合缓冲液中预稀释,随后添加至等体积的硫醇化抗体中。
在室温和搅拌下将缀合反应混合物孵育2 h。在孵育后,将缀合反应混合物离心并经由0.2 μm过滤器过滤。缀合物的纯化可经由层析使用多种方法实现。在一种方法中,在使用含有5 mM甘氨酸和150 mM NaCl的50 mM pH 7.2 HEPES缓冲液预平衡的SEPHACRYL™ S200柱上使用大小排阻层析来纯化缀合物。以28 cm/h的线性流速实施层析。收集、合并并浓缩含有缀合物的级分。在替代方法中,可经由离子交换层析实现纯化。条件随抗体而变化且应在每一情形下进行优化。例如,将抗体-药物缀合物反应混合物施加至在含有5 mM甘氨酸的50 mM pH 5.5 HEPES预平衡的SP-SEPHAROSE™柱中。使用0-1 M NaCl于pH 5.5平衡缓冲液中的梯度来洗脱抗体缀合物。合并含有缀合物的相关级分并针对配制缓冲液(含有5 mM甘氨酸和100 mM NaCl的50 mM pH 7.2 HEPES缓冲液)进行透析。
本领域技术人员应理解,上述条件和方法是示例性和非限制性的,且其它缀合方法是本领域已知的并且可用于本发明中。
通过上述程序制得的缀合物由式(II-1)代表。其是化合物(III-1)和抗间皮素抗体6A4的缀合物(Terrett等人,2012):
。
本领域技术人员应理解,此类缀合物制剂可具有不同取代比(通常范围为1至5)的部分,且此类制剂可由式(II-1')代表:
其中R6是Me或n-Pr且Ab是抗体。抗体优选是抗CD70、抗间皮素或抗磷脂酰肌醇蛋白聚糖-3抗体。
药物组合物
在另一方面,本公开内容提供药物组合物,其包含本发明化合物或其缀合物,其与药学上可接受的载体或赋形剂一起配制。其可任选地含有一种或多种其它药学活性成分,例如抗体或另一药物。药物组合物可在组合治疗中与另一治疗剂、尤其另一抗癌剂一起施用。
药物组合物可包含一种或多种赋形剂。可使用的赋形剂包括载体、表面活性剂、增稠剂或乳化剂、固体粘合剂、分散或悬浮助剂、增溶剂、染色剂、矫味剂、包衣剂、崩解剂、润滑剂、甜味剂、防腐剂、等渗剂和其组合。合适赋形剂的选择和应用教导于Gennaro编辑,Remington: The Science and Practice of Pharmacy,第20版(Lippincott Williams & Wilkins 2003)中,其公开内容通过引用并入本文。
优选地,药物组合物适于静脉内施用、肌内施用、皮下施用、肠胃外施用、脊椎或表皮施用(例如通过注射或输注)。取决于施用途径,可将活性化合物包被于材料中保护其免受酸作用和其它可使其失活的天然条件影响。短语“肠胃外施用”意指除经肠和局部施用外的施用模式,通常通过注射且包括但不限于静脉内、肌内、动脉内、鞘内、囊内、眶内、心脏内、真皮内、腹膜内、经气管、皮下、表皮下、关节内、囊下、蛛网膜下、脊柱内、硬膜外和胸骨内注射和输注。或者,药物组合物可经由非肠胃外用途径施用,例如局部、表皮或粘膜施用途径,例如鼻内、经口、阴道、直肠、舌下或局部。
药物组合物可呈无菌水溶液或分散液的形式。其也可配制于微乳液、脂质体或其它适用于实现高药物浓度的有序结构中。组合物也可以冻干物的形式提供,用于在施用之前于水中重构。
可与载体材料组合产生单一剂型的活性成分的量将取决于所治疗受试者和特定施用模式而变化,且通常是产生治疗性效应的组合物的量。通常,在100个百分比中,该量的范围为约0.01%至约99%活性成分、优选地约0.1%至约70%活性成分、最优选地约1%至约30%活性成分,其与药学上可接受的载体组合。
调节剂量方案以提供治疗反应。例如,可施用单一浓注,可经一段时间施用几个分开剂量或可如状况紧急程度所示按比例减少或增加剂量。以剂量单元形式来配制肠胃外组合物尤其有利于方便施用和剂量一致性。“剂量单位形式”是指适合作为单位剂量用于待治疗受试者的物理分散单位;每一单位含有预定量的活性化合物,该预定量经计算以便与所需药物载体组合产生期望治疗反应。
剂量范围为约0.0001 mg/kg宿主体重至100 mg/kg宿主体重,更通常0.01 mg/kg宿主体重至5 mg/kg宿主体重。例如,剂量可以是0.3 mg/kg体重、1 mg/kg体重、3 mg/kg体重、5 mg/kg体重或10 mg/kg体重或在1-10 mg/kg范围内。示例性治疗方案以以下方式施用:每周一次、每两周一次、每三周一次、每四周一次、每月一次、每3个月一次或每3至6个月一次。优选剂量方案包括经由静脉内施用来施用1 mg/kg体重或3 mg/kg体重,其使用下列给药时间表之一:(i)每四周给予6个剂量,然后每三个月一次;(ii)每三周一次;(iii)每三周给予一次3 mg/kg体重、随后1 mg/kg体重。在一些方法中,调节剂量以实现约1-1000 μg /ml和在一些方法中约25-300 μg /ml的血浆抗体浓度。
本发明化合物的“治疗有效量”优选地导致降低疾病症状的严重度,增加疾病无症状期的频率和持续时间,或预防由疾病折磨导致的损害或失能。例如,对于携带肿瘤的受试者的治疗而言,“治疗有效量”优选地相对于未治疗受试者将肿瘤生长抑制至少约20%、更优选地至少约40%、甚至更优选地至少约60%且更优选地至少约80%。治疗有效量的治疗化合物可降低受试者的肿瘤大小或以其它方式改善症状,该受试者通常是人,但可以是另一哺乳动物。
药物组合物可以是受控释放或持续释放的制剂,包含植入物、经皮贴剂和微囊封递送系统。可使用生物可降解的生物相容性聚合物,例如乙烯基乙酸乙烯酯、聚酐、聚乙醇酸、胶原、聚原酸酯和聚乳酸。参见例如Sustained and Controlled Release Drug Delivery Systems, J.R. Robinson编辑,Marcel Dekker公司,New York, 1978。
可经由医学装置施用治疗性组合物,例如(1)无针皮下注射装置(例如US 5,399,163、5,383,851、5,312,335、5,064,413、4,941,880、4,790,824和4,596,556);(2)微输注泵(US 4,487,603);(3)经皮装置(US 4,486,194);(4)输注设备(US 4,447,233和4,447,224);和(5)渗透装置(US 4,439,196和4,475,196);其公开内容通过引用并入本文。
在某些实施方案中,可配制药物组合物以确保在体内合理分布。例如,为了确保本发明的治疗性化合物穿过血-脑屏障,可将其配制于脂质体中,所述脂质体可另外包含靶向部分以增强至特点细胞或器官的选择性转运。参见例如US 4,522,811;5,374,548;5,416,016;和5,399,331;V.V. Ranade (1989) J. Clin. Pharmacol. 29:685;Umezawa等人,(1988) Biochem. Biophys. Res. Commun. 153:1038;Bloeman 等人,(1995) FEBS Lett.357:140;M. Owais等人,(1995) Antimicrob. Agents Chemother. 39:180;Briscoe等人,(1995) Am. J. Physiol. 1233:134;Schreier等人,(1994) J. Biol. Chem. 269:9090;Keinanen和Laukkanen (1994) FEBS Lett. 346:123;以及Killion和Fidler (1994) Immunomethods4:273。
用途
本发明化合物或其缀合物可用于治疗疾病,例如,但不限于,过度增殖性疾病,包括:头颈癌,包括头部肿瘤、颈部肿瘤、鼻腔肿瘤、副鼻窦肿瘤、鼻咽肿瘤、口腔肿瘤、口咽肿瘤、喉部肿瘤、下咽部肿瘤、唾液腺肿瘤和副神经节瘤;肝癌和胆管树癌,具体而言肝细胞癌;肠癌,具体而言结肠直肠癌;卵巢癌;小细胞和非小细胞肺癌(SCLC和NSCLC);乳癌肉瘤,例如纤维肉瘤、恶性纤维组织细胞瘤、胚胎型横纹肌肉瘤、平滑肌肉瘤、神经纤维肉瘤、骨肉瘤、滑膜肉瘤、脂肪肉瘤和肺泡状软组织肉瘤;白血病,例如急性前髓细胞性白血病(APL)、急性骨髓性白血病(AML)、急性淋巴胚细胞白血病(ALL)和慢性骨髓性白血病(CML);中枢神经系统赘瘤,具体而言脑癌;多发性骨髓瘤(MM);淋巴瘤,例如霍奇金氏淋巴瘤(Hodgkin's lymphoma)、淋巴浆细胞样淋巴瘤、滤泡淋巴瘤、粘膜相关淋巴样组织淋巴瘤、套细胞淋巴瘤、B-谱系大细胞淋巴瘤、伯基特氏淋巴瘤(Burkitt’s lymphoma)和T细胞间变性大细胞淋巴瘤。临床上,本文所述组合物的方法和用途的实践将导致减小癌性生长的大小或数量和/或减轻相关症状(当适用时)。病理上,本文所述组合物的方法和用途的实践可产生病理上相关反应,例如:抑制癌细胞增殖、减小癌症或肿瘤的大小、预防进一步转移和抑制肿瘤血管生成。治疗此类疾病的方法包括向受试者施用治疗有效量的发明组合。可根据需要重复该方法。尤其而言,癌症可以是肾癌、肺癌、胃癌或卵巢癌。
本发明化合物或其缀合物可与其它治疗剂组合施用,所述治疗剂包括抗体、烷基化剂、血管生成抑制剂、抗代谢物、DNA裂解剂、DNA交联剂、DNA嵌入剂、DNA小沟结合剂、烯二炔、热休克蛋白90抑制剂、组蛋白去乙酰酶抑制剂、免疫调节剂、微管稳定剂、核苷(嘌呤或嘧啶)类似物、出核转运抑制剂、蛋白酶体抑制剂、拓扑异构酶(I或II)抑制剂、酪氨酸激酶抑制剂和丝氨酸/苏氨酸激酶抑制剂。具体治疗剂包括阿达木单抗(adalimumab)、安丝菌素P3 (ansamitocin P3)、auristatin、苯达莫司汀(bendamustine)、贝伐珠单抗(bevacizumab)、比卡鲁胺(bicalutamide)、博来霉素(bleomycin)、硼替佐米(bortezomib)、白消安(busulfan)、卡利斯他汀A (callistatin A)、喜树碱(camptothecin)、卡培他滨(capecitabine)、卡铂(carboplatin)、卡莫司汀(carmustine)、西妥昔单抗(cetuximab)、顺铂(cisplatin)、克拉屈滨(cladribin)、阿糖胞苷(cytarabin)、念珠藻素、达卡巴嗪(dacarbazine)、达沙替尼(dasatinib)、道诺霉素(daunorubicin)、多西他赛(docetaxel)、多柔比星(doxorubicin)、多卡米星(duocarmycin)、达内霉素A (dynemycin A)、埃博霉素、依托泊苷(etoposide)、氟尿苷(floxuridine)、氟达拉滨(fludarabine)、5-氟尿嘧啶(5-fluorouracil)、吉非替尼(gefitinib)、吉西他滨(gemcitabine)、易普利姆玛(ipilimumab)、羟基脲(hydroxyurea)、伊马替尼(imatinib)、英利昔单抗(infliximab)、干扰素、白介素、β-拉帕醌(β-lapachone)、来那度胺(lenalidomide)、伊立替康(irinotecan)、美登素、氮芥(mechlorethamine)、美法仑(melphalan)、6-巯基嘌呤、氨甲蝶呤(methotrexate)、丝裂霉素C (mitomycin C)、尼罗替尼(nilotinib)、奥沙利铂(oxaliplatin)、紫杉醇、丙卡巴肼(procarbazine)、辛二酰基苯胺异羟肟酸(SAHA)、6-硫鸟嘌呤、塞替派(thiotepa)、替尼泊苷(teniposide)、托泊替康(topotecan)、曲妥珠单抗(trastuzumab)、曲古抑菌素A (trichostatin A)、长春碱、长春新碱(vincristine)和长春地辛(vindesine)。
实施例
本发明的实践可通过参照下列实施例来进一步理解,所述实施例通过说明方式提供且不具有限制性。
实施例1-化合物(III-1)
本实施例描述化合物(III-1)的合成,相应方案组合显示于图1、2a-2b和3中。
化合物2. 将甲苯(150 mL)中的化合物1 (6 g,16.6 mmol;根据Peltier等人,2006制备)和多聚甲醛(9.94 g, 331 mmol)的混合物在70℃下于密封器皿中加热24 h。薄层层析(TLC)显示反应完成。经由硅藻土™ (CELITE™)过滤介质过滤反应混合物且使用甲苯充分洗涤滤饼。在蒸发溶剂之后,通过快速层析(使用二氯甲烷(DCM)中的0-70%乙酸乙酯(EtOAc)的梯度从硅胶洗脱)纯化粗产物以提供4.76 g作为浅黄色油状物的化合物2。MS: (+) m/z 375.2 (M+1)。
化合物3. 在结合载体的氰基硼氢化物(MP-BH3CN)树脂(4.85 g, 12.7 mmol)存在下,将盐酸(4.0 M,1,4-二氧杂环己烷中,12.24 mL,50.8 mmol)逐滴添加至乙腈(62 mL)和甲醇(6.8 mL)中的化合物2 (4.76 g, 12.7 mmol)的溶液中。将反应混合物在室温(RT)下搅拌3 h。LCMS显示反应完成。过滤掉树脂并使用乙腈-甲醇混合物洗涤。在蒸发溶剂之后,通过快速层析(从使用DCM中且含有1% NH4OH的0-10%甲醇的梯度的硅胶洗脱)纯化粗产物以提供粗制化合物3。
浓缩产物级分,使用EtOAc稀释,并使用NaHCO3饱和水溶液洗涤一次以去除过量铵盐。使用EtOAc将水性级分反萃取一次。干燥合并的有机相并浓缩以提供2.82 g作为发泡固体的化合物3。MS: (+) m/z 273.2 (M+1)。
化合物4. 在0℃下,将聚合物结合的N-苄基-N-环己基碳化二亚胺(Aldrich, 4.5 g, 5.21 mmol)添加至DCM (48 mL)中的化合物3 (1.42 g, 5.21 mmol)、叔丁醇(0.72 g, 5.32 mmol)和Boc保护的异亮氨酸3a (1.27 g, 5.47 mmol)的溶液中。将反应混合物在室温下搅拌过夜。过滤掉树脂并使用DCM洗涤。浓缩滤液,使用EtOAc稀释,并使用NaHCO3饱和水溶液洗涤一次。使用EtOAc将水溶液萃取两次。干燥合并的有机层,过滤并浓缩。通过快速层析(从使用DCM中且含有1% NH4OH的0-10%甲醇的梯度的硅胶洗脱)纯化粗产物以提供含有中间体产物3b的级分。
合并含有产物的级分,浓缩,使用EtOAc稀释,并使用NaHCO3饱和水溶液洗涤以去除过量铵盐。使用EtOAc将水性级分反萃取一次。干燥合并的有机相并浓缩以提供作为白色固体的中间体产物3b。
将甲苯(50 mL)中的中间体产物3b在搅拌下于密封器皿中加热至90℃过夜。LCMS显示反应完成。蒸发溶剂。通过快速层析(从使用己烷中的0-100% EtOAc的梯度的硅胶洗脱)纯化粗产物以提供1.3 g作为浅黄色固体的化合物4。MS: (+) m/z 486.3 (M+1)。
化合物5. 将三氟乙酸(TFA, 26 mL)添加至DCM (26 mL)中的化合物4的混合物中。在室温下搅拌30 min之后,LCMS显示反应完成。浓缩溶液,使用EtOAc稀释,并使用NaHCO3饱和水溶液洗涤一次。使用EtOAc将水溶液反萃取两次。干燥合并的有机层,过滤,并浓缩以提供1.03 g作为白色固体的化合物5。MS: (+) m/z 386.3 (M+1)。
化合物6. 在0℃下,将DCC (0.664 g, 3.22 mmol)添加至DCM中的化合物5 (1.03 g, 2.68 mmol)、(R)-1-甲基哌啶-2-甲酸5a (0.4 g,2.81 mmol;根据Peltier等人,2006制备)和叔丁醇(0.369 g, 2.73 mmol)的混合物中。将反应混合物升温至室温并在室温下搅拌过夜。过滤掉固体,且浓缩滤液。将残余物溶于EtOAc中并使用NaHCO3饱和水溶液洗涤一次。使用EtOAc将水溶液反萃取两次。干燥合并的有机层,过滤,并浓缩。通过快速层析(从使用DCM中的0-20%甲醇的梯度的硅胶洗脱)纯化粗产物以提供1.23 g作为浅黄色固体的化合物6。MS: (+) m/z 511.4 (M+1)。
化合物7. 将N,N-二甲基甲酰胺(DMF, 10 mL)中的N,N-二异丙基乙胺(DIEA,也称为DIPEA,0.972 mL,5.58 mmol)、碳酸双(4-硝基苯基)酯(BNPC, 1.698 g, 5.58 mmol)和化合物6 (0.57 g, 1.116 mmol)的混合物在室温下搅拌过夜。LCMS显示反应完成。蒸发溶剂。通过硅胶快速层析(使用DCM中的0-20%甲醇的梯度)纯化粗产物以提供0.68 g作为黄色油状物的化合物7。MS: (+) m/z 676.4 (M+1)。
化合物8. 将甲醇中的甲胺(2.0 M, 0.089 mL, 0.178 mmol)添加至甲醇(1 mL)中的化合物7 (0.1 g, 0.148 mmol)中。在室温下将反应混合物搅拌10 min之后,LCMS显示反应完成。蒸发溶剂以提供0.084 g化合物8。MS: (+) m/z 568.4 (M+1)。
化合物9. 在室温下,将水(0.5 mL)中的氢氧化锂(7.09 mg, 0.296 mmol)添加至1,4-二氧杂环己烷(0.5 mL)中的化合物8 (0.084 g, 0.148 mmol)的溶液中。在室温下将反应混合物搅拌2 h之后,LCMS显示反应完成。蒸发溶剂。通过快速层析(从使用DCM中的0-30%甲醇的梯度的硅胶洗脱)纯化粗产物以提供0.075 g作为白色固体的化合物9。MS: (+) m/z 554.4 (M+1)。
化合物10. 在0℃下,将三乙胺(11.73 mL, 84 mmol)添加至乙腈(300 mL)中的二碳酸二-叔丁酯(BOC2O, 10.57 mL, 46.0 mmol)和(S)-2-氨基-3-(4-硝基苯基)丙酸甲酯盐酸盐9a (10 g, 38.4 mmol)的混合物中。将反应混合物升温至室温,并在室温下搅拌过夜。LCMS显示反应完成。浓缩反应混合物,且将产物再溶于200 mL二乙醚中。过滤掉固体,且浓缩滤液。通过快速层析(从使用己烷中的0-50% EtOAc的梯度的硅胶洗脱)纯化粗产物以提供11.3 g作为白色固体的Boc保护中间体。
将Pd/C催化剂(10 wt.%, 0.85 g, 7.99 mmol)添加至MeOH (200 mL)中的Boc保护中间体(15 g, 46.2 mmol)的溶液中。在氢气氛下将反应混合物搅拌过夜。过滤Pd/C催化剂,且浓缩滤液以提供13.6 g作为白色固体的化合物10。MS: (+) m/z 195.2 (M+1-Boc)。
化合物11. 在0℃下,将吡啶(5.77 mL, 71.3 mmol)添加至DCM (185 mL)中的氯甲酸苄酯(10.18 mL, 71.3 mmol)和化合物10 (17.5 g, 59.5 mmol)的溶液中。将反应混合物升温至室温且在室温下搅拌过夜。通过添加NaHCO3饱和水溶液来猝灭反应,并使用盐水洗涤。干燥有机层,过滤,并浓缩。通过快速层析(从使用己烷中的0-50% EtOAc的梯度的硅胶洗脱)纯化粗产物以提供22.6 g作为无色油状物的化合物11。MS: (+) m/z 329.2 (M+1-Boc)。
化合物12. 在-78℃下,将己烷中的二异丁基氢化铝(DIBAL-H) (1 M, 26.5 mL, 26.5 mmol)添加至DCM (39 mL)中的化合物11 (5.17 g, 12.07 mmol)的溶液中。将反应混合物在-78℃下搅拌2 h。在-78℃下添加乙酸(24 mL)和甲苯(36 mL)。将反应混合物升温至室温。将酒石酸(10%水溶液,69 mL)添加至反应混合物中。使用己烷和EtOAc (v/v 1:1)混合物萃取水溶液。干燥合并的有机层,过滤,并浓缩。通过快速层析(从使用己烷中的0-50% EtOAc的梯度的硅胶洗脱)纯化粗产物以提供3.12 g作为白色固体的化合物12。MS: (+) m/z 299.2 (M+1-Boc)。
化合物13. 在0℃下,将二丁基(((三氟甲基)磺酰基)氧基)硼烷(Bu2BOTf,1 M,DCM中,8.61 mL,8.61 mmol)和DIEA (1.637 mL, 9.40 mmol)添加至DCM (7.8 mL)中的(S)-4-异丙基-3-丙酰基噁唑烷-2-酮12a (1.450 g, 7.83 mmol)的溶液中。将反应混合物在0℃下搅拌45 min。在-78℃下,将DCM (7.8 mL)中的化合物12 (3.12 g, 7.83 mmol)的溶液添加至反应混合物中。将反应混合物升温至室温过夜。添加磷酸钠缓冲液(pH 7, 29 mL)。使用DCM萃取水溶液。使用盐水洗涤合并的有机层,干燥,过滤,并浓缩。
将残余物再溶于甲醇(130 mL)中并冷却至0℃。在0℃下,将H2O2水溶液(30%, 39.7 mL)添加至反应混合物中。将反应混合物在0℃下搅拌4 h。添加水(39 mL)。蒸发一些溶剂(MeOH)。使用EtOAc萃取水溶液。使用5% NaHCO3溶液和盐水洗涤合并的有机层,干燥,过滤,并浓缩。通过快速层析(从使用己烷中的0-50% EtOAc的梯度的硅胶洗脱)纯化粗产物以提供4.23 g作为无色油状物的化合物13。MS: (+) m/z 484.3 (M+1-Boc)。
化合物14. 将二(1H-咪唑-1-基)甲硫酮(1.5 g, 8.42 mmol)添加至THF (20 mL)中的化合物13 (2.46 g, 4.21 mmol)的溶液中。将反应混合物回流过夜。LCMS显示反应完成。蒸发溶剂。通过硅胶快速层析(使用己烷中的0-50% EtOAc的梯度)纯化粗产物以提供1.25 g作为白色固体的化合物14。MS: (+) m/z 694.3 (M+1)。
化合物15. 将(E)-2,2'-(二氮烯-1,2-二基)双(2-甲基丙腈) (AIBN, 0.016 g, 0.095 mmol)添加至化合物14 (1.78 g, 2.57 mmol)和三丁基锡烷(Bu3SnH, 1.380 mL, 5.13 mmol)的溶液中。将反应混合物回流30 min (142℃下的油浴温度)。蒸发溶剂。通过快速层析(从使用己烷中的0-33% EtOAc的梯度的硅胶洗脱)纯化粗产物以提供0.84 g作为浅黄色油状物的化合物15。MS: (+) m/z 468.3 (M+1-Boc)。
化合物16. 在0℃下,将水(3.7 mL)中的LiOH (0.071 g, 2.96 mmol)添加至四氢呋喃(THF, 11.4 mL)中的化合物15 (0.84 g, 1.480 mmol)的溶液中,随后添加30% H2O2水溶液(0.271 mL, 8.88 mmol)。在0℃下将反应混合物搅拌4 h之后,添加20 mL 1.33 M Na2SO3水溶液以猝灭反应。添加盐酸(1 M)以将pH调节至2-3。使用DCM萃取所得水溶液。干燥合并的有机层,过滤,并浓缩。通过快速层析(从使用己烷中的0-75% EtOAc的梯度的硅胶洗脱)纯化粗产物以提供0.53 g作为无色油状物的化合物16。MS: (+) m/z 357.3 (M+1-Boc)。
化合物17. 将浓盐酸(4滴)添加至甲醇(17.7 mL)中的2,2-二甲氧基丙烷(3.53 mL, 28.7 mmol)和化合物16 (0.53 g, 1.161 mmol)的溶液中。将反应混合物在室温下搅拌过夜。LCMS显示反应完成。LCMS还显示形成一些去保护副产物16a。蒸发溶剂。
在室温下,将三乙胺(2.2当量,0.36 mL)添加至乙腈中的上述残余物和BOC2O (1.2当量,304.3 mg)的溶液中以再保护副产物16a。将反应混合物在室温下搅拌2 h。LCMS显示反应完成。蒸发溶剂。添加水(7 mL),且使用EtOAc萃取水溶液。干燥合并的有机层,过滤,并浓缩。通过快速层析(从使用己烷中的0-50% EtOAc的梯度的硅胶洗脱)纯化粗产物以提供0.3 g作为无色油状物的化合物17。MS: (+) m/z 371.3 (M+1-Boc)。
化合物18. 将甲醇(6 mL)中的化合物17 (0.223 g, 0.474 mmol)和Pd/C 10 wt% (20 mg, 0.474 mmol)的混合物在H2下搅拌过夜。过滤掉Pd/C催化剂,并浓缩滤液。通过快速层析(从使用己烷中的0-50% EtOAc的梯度的硅胶洗脱)纯化粗产物以提供0.112 g作为白色固体的化合物18。MS: (+) m/z 237.2 (M+1-Boc)。
化合物19. 将DMF (12.4 mL)中的化合物18 (0.204 g, 0.606 mmol)、N-乙基-N′-(3-二甲基氨基丙基)碳化二亚胺盐酸盐(EDC, 0.174 g, 0.910 mmol)和Fmoc保护的瓜氨酸18a (0.361 g, 0.910 mmol)的混合物在室温下搅拌过夜。添加饱和NH4Cl溶液(20 mL)以猝灭反应。使用EtOAc萃取水溶液。干燥合并的有机层,过滤,并浓缩。通过硅胶快速层析(使用DCM中的0-30% MeOH的梯度)纯化粗产物以提供0.25 g作为白色固体的化合物19。MS: (+) m/z 716.4 (M+1)。
化合物20. 将哌啶(0.5 mL, 5.06 mmol)添加至DMF (5 mL)中的化合物19 (0.25 g, 0.349 mmol)的溶液中。在将反应混合物在室温下搅拌20 min之后,蒸发溶剂以提供作为残余物的Fmoc去保护中间体。
将DIEA添加至DMF (2 mL)中的(S)-2-((((9H-茀-9-基)甲氧基)羰基)氨基)-3-甲基丁酸19a (0.142 g, 0.418 mmol)和六氟磷酸N,N,N′,N′-四甲基-O-(7-氮杂苯并三唑-1-基)脲鎓(HATU, 0.146 g, 0.383 mmol)的溶液中,从而将pH调节至8-9。在将反应混合物在室温下搅拌5 min之后,将DMF (1 mL)中的上述残余物和DIEA添加至反应混合物中,从而将pH调节至8-9。在将反应混合物在室温下搅拌15 min之后,添加20 mL含有8 mL 0.1% TFA水的水。使用EtOAc萃取水溶液。干燥合并的有机层,过滤,并浓缩。通过快速层析(从使用DCM中的0-20% MeOH的梯度的硅胶洗脱)纯化粗产物以提供0.24 g作为白色固体的化合物20。MS: (+) m/z 815.4 (M+1)。
化合物21. 将哌啶(0.3 mL)添加至DMF (3 mL)中的化合物20的溶液中。将反应混合物在室温下搅拌1 h。LCMS显示反应完成。蒸发溶剂。
将水(2 mL)中的氢氧化锂(0.028 g, 1.176 mmol)添加至THF (4 mL)中的上述残余物的溶液中。在将反应混合物在室温下搅拌4 h之后,添加HCl水溶液(0.1N)以酸化反应混合物(pH 2-3)。部分地蒸发溶剂,并冻干以提供作为白色固体的化合物21。MS: (+) m/z 579.4 (M+1)。
化合物22. 将DIEA添加至DMF (3 mL)中的ε-马来酰亚胺基己酸N-羟基琥珀酰亚胺酯21a (Tokyo Chemical Industry, 64.7 mg, 0.210 mmol)和化合物21 (81 mg, 0.14 mmol)的混合物中,从而调节pH 8-9。在将反应混合物在室温下搅拌2 h之后,添加10 mL乙腈和含有0.1% TFA的水的1:1 (v/v)混合物。通过制备型高效液相层析(HPLC)纯化产物22。MS: (+) m/z 772.5 (M+1)。
化合物23. 在室温下,将2,2,2-三氟乙酸(0.7 mL, 0.013 mmol)添加至DCM (1 mL)中的化合物22 (30 mg, 0.039 mmol)的混合物中。在将反应混合物在室温下搅拌10 min之后,LCMS显示反应完成。蒸发溶剂,从而提供化合物23。MS: (+) m/z 672.4 (M+1)。
化合物(III-1). 将DIEA添加至DMF (1 mL)中的化合物9 (23.66 mg, 0.043 mmol)和HATU (14.77 mg, 0.039 mmol)的溶液中。将反应混合物的pH调节至8-9。在将反应混合物在室温下搅拌10 min之后,添加DMF (1 mL)中的化合物23 (26.1 mg, 0.039 mmol)和DIEA。将反应溶液的pH调节至8-9。在将反应混合物在室温下搅拌10 min之后,LCMS显示反应完成。通过添加10 mL含有0.1% TFA的水和乙腈的1:1 (v/v)混合物来猝灭反应。通过制备型HPLC纯化产物化合物(III-1)。MS: (+) m/z 1207.7 (M+1)。
可使用具有马来酰亚胺基的化合物(例如(III-1))通过与抗体或其它配体上的巯基进行反应来制备缀合物。巯基可以是来自半胱氨酸残基的巯基或通过使用2-亚氨基硫杂环戊烷衍生赖氨酸残基获得的巯基。
实施例2 –化合物(III-2)
本实施例描述化合物(III-2)的合成,相应方案显示于图4中。
化合物24. 在室温下,将N-乙基-N-异丙基丙烷-2-胺(0.556 mL, 3.19 mmol)添加至DMF (5 mL)中的甘氨酸叔丁酯盐酸盐23a (0.209 g, 1.596 mmol)、Fmoc-氨基氧基乙酸23b (0.5g, 1.596 mmol)和HATU (0.607 g, 1.596 mmol)的溶液中。在将反应混合物在室温下搅拌1 h之后,添加0.1% TFA水溶液(20 mL)。使用EtOAc萃取水溶液,且干燥合并的有机层,过滤,并浓缩。通过快速层析(从使用己烷中的0-70% EtOAc的梯度的硅胶洗脱)纯化粗产物以提供0.45 g作为无色油状物的化合物24。MS: (+) m/z 449.2 (M+23)。
化合物25. 在室温下,将TFA (3 mL, 1.437 mmol)添加至DCM (0.5 mL)中的化合物24 (0.45 g, 1.055 mmol)的溶液中。将反应混合物在室温下搅拌过夜。蒸发溶剂。通过快速层析(从使用DCM中的0-30% MeOH的梯度的硅胶洗脱)纯化粗产物以提供0.39 g作为白色固体的化合物25。MS: (+) m/z 371.1 (M+1)。
化合物26. 在室温下,将N,N'-甲烷二亚基二环己胺(DCC, 0.261 g, 1.267 mmol)添加至DCM (6 mL)中的化合物25和1-羟基吡咯烷-2,5-二酮(0.146 g, 1.267 mmol)的溶液中。在将反应混合物在室温下搅拌过夜之后,过滤掉固体。然后浓缩滤液。通过快速层析(从使用己烷中的0-100% EtOAc的梯度的硅胶洗脱)纯化粗产物以提供0.43 g作为无色油状物的化合物26。
化合物27. 在室温下,将DIEA添加至DMF中的化合物21 (50 mg, 0.086 mmol)和化合物26 (60.6 mg, 0.130 mmol)的溶液中,从而将pH调节至8-9。在将反应混合物在室温下搅拌4 h之后,通过添加10 mL 0.1%TFA水溶液和乙腈的1:1混合物来猝灭反应。制备型HPLC纯化提供55 mg作为白色固体的化合物27,MS: (+) m/z 931.4 (M+1)。
化合物28. 在室温下,将TFA (1 mL, 0.059 mmol)添加至DCM (2 mL)中的化合物27 (55 mg, 0.059 mmol)的溶液中。将反应混合物在室温下搅拌10 min。蒸发溶剂。
将DIEA添加至DMF (1 mL)中的化合物9 (32.7 mg, 0.059 mmol)和HATU (22.47 mg, 0.059 mmol)的溶液中。将反应混合物的pH调节至8-9。在将反应混合物在室温下搅拌10 min之后,添加DMF (2 mL)和DIEA。将反应溶液的pH调节至8-9。在将反应混合物在室温下搅拌10 min之后,LCMS显示反应完成。通过添加20 mL含有0.1% TFA的水和乙腈的1:1 (v/v)混合物来猝灭反应。制备型HPLC纯化提供70 mg作为白色固体的化合物28。MS: (+) m/z 684.1 (M/2+1)。
化合物(III-2). 将哌啶添加至DMF (4 mL)中的化合物28 (70 mg, 0.051 mmol)的溶液中。在将反应混合物在室温下搅拌20 min之后,添加乙腈和0.1% TFA水溶液的1:1混合物(40 mL)。制备型HPLC纯化提供46 mg作为白色固体的化合物(III-2)。MS: (+) m/z 1144.6 (M+1)。
可使用具有羟胺基团的化合物(例如(III-2))与抗体或具有醛或酮官能团的其它配体(例如)通过并入非天然氨基酸4-乙酰基苯丙氨酸来形成缀合物。
实施例3 -化合物(I-2)和(I-3)
化合物(I-2)和(I-3)的合成示意性显示于图5中。
化合物(I-3).将DIEA添加至DMF (0.3 mL)中的化合物9 (10 mg, 0.018 mmol)和HATU (6.87 mg, 0.018 mmol)的溶液中,从而将pH调节至8-9。在将反应混合物在室温下搅拌10 min之后,添加DMF (0.5 mL)和DIEA中的化合物29 (根据Cheng等人,2011,实施例17制备;4.56 mg,0.018 mmol),从而将pH调节至8-9。在将反应混合物在室温下搅拌20 min之后,通过添加4 mL乙腈和0.1% TFA水溶液的1:1混合物来猝灭反应。制备型HPLC纯化提供12 mg作为白色固体的化合物(I-3) (I-2)。MS: (+) m/z 788.4 (M+1)。
化合物(I-2).将甲醇(0.5 mL)中的化合物(I-3) (12 mg, 0.015 mmol)和10 wt% Pd/C (4 mg, 0.015 mmol)的混合物在H2气氛下搅拌过夜。过滤掉催化剂,且浓缩滤液。制备型HPLC纯化提供8.1 mg作为白色固体的化合物(I-2)。MS: (+) m/z 758.4 (M+1)。
可以类似地通过使用在α-甲基位置具有混合立体化学的化合物代替化合物29来制备化合物(I-1) (Cheng等人,2011)。
实施例4 -化合物(III-4)
图6a至6c示意性组合显示化合物(III-4)的合成。
化合物32. 将化合物30 (Aldrich, 3.5g, 13.9 mmol)溶于50 mL DCM中。在5℃下,向该溶液中添加戴斯-马丁过碘烷(Dess-Martin periodinane) (11.8 g, 27.9 mmol)。在10 min之后,将混合物升温至室温。再过一小时之后,使用NaHCO3饱和水溶液和NaS2O3饱和水溶液猝灭反应。在使用乙醚萃取之后,使用NaHCO3水溶液和然后盐水洗涤乙醚萃取物且干燥并蒸发成粘性油状物。将油状物溶于50 mL DCM中,向其中添加市售化合物31 (5.05 g, 13.93 mmol)。在10 min之后,将反应混合物溶于EtOAc中,使用NaHCO3水溶液和然后盐水洗涤并干燥,过滤并蒸发溶剂。在柱层析(EtOAc:己烷,0-20%梯度)之后,获得作为白色固体的化合物32 (2.3 g,6.90 mmol,产率为49.5%)。其具有与文献一致的NMR光谱(Wipf等人,2004a)。
化合物33. 在5℃下,将盐酸(7.80 mL,31.2 mmol,4 M,二氧杂环己烷中)添加至化合物32 (5.2 g, 15.60 mmol)的DCM溶液中。在去保护反应完成之后,蒸发反应混合物且获得作为白色固体的化合物33 (4.21 g,15.60 mmol,产率为100%,盐酸盐),其未经进一步纯化即用于下一步骤中。
化合物35. 在5℃下,向20 mL DMF中的化合物34 (根据Sani等人,2007制备,4.00 g,11.6 mmol)的溶液中添加HATU (4.61 g, 12.12 mmol)和DIPEA (6 ml, 34.4 mmol)。在10 min之后,添加化合物33 (2.71 g, 11.60 mmol)。再过半小时之后,将混合物溶于EtOAc中,其使用10%柠檬酸水溶液、NaHCO3饱和水溶液和盐水洗涤。在干燥且过滤之后,蒸发有机相以得到作为油状物的化合物35 (6.49 g,11.60 mmol,产率为100%,[M+Na]+,计算值:582.3,实验值:582.3),其未经进一步纯化即用于下一步骤中。
化合物36. 在5℃下,将NaBH4 (4.66 g, 123 mmol)逐份添加至化合物35 (6.49 g, 11.6 mmol)和NiSO4(H2O)6 (6.48 g, 24.66 mmol)的100 mL甲醇溶液中。(注意:生成氢。)在30 min之后,添加NaHCO3饱和水溶液,随后添加EtOAc。在经由硅藻土™过滤之后,使有机相与水相分离,使用盐水洗涤,干燥,过滤并蒸发以得到化合物36 (5.6 g,9.97 mmol,产率为81%,[M+1]+,计算值:562.3,实验值:562.4),其未经进一步纯化即用于下一步骤中。
化合物37. 在5℃下,将化合物36 (5.6 g, 9.97 mmol)溶于30 mL吡啶中。将乙酸酐(4 g, 39.2 mmol)添加至该溶液中。在10 min之后,将混合物升温至室温。在约一小时之后,浓缩反应混合物。将所得残余物溶于EtOAC中且依次使用10%柠檬酸水溶液、NaHCO3饱和水溶液和盐水洗涤有机相。干燥有机相,过滤并浓缩以得到化合物37 (5.8 g,9.61 mmol,产率为100%,[M+1]+,计算值:604.3,实验值:604.4),其未经进一步纯化即用于下一步骤中。
化合物38a和38b. 在-78℃下,将化合物37 (0.8 g, 1.3 mmol)溶于5 mL甲醇中。向该溶液中添加NaOMe (331 uL,1.33 mmol,4 M,MeOH中)。经1 hr将混合物升温至室温。将混合物溶于EtOAc中,使用10%柠檬酸水溶液、NaHCO3饱和水溶液和盐水洗涤。干燥分离的有机相,过滤并蒸发以得到乙酯和甲酯的混合物(分别是化合物38a和38b)。在随后几个步骤期间不分离酯混合物,直至二者在后一步骤中水解成羧酸。
化合物40a和40b.将来自上述反应的化合物38a和38b的混合物溶于20 mL DCM中。在5℃下,向该溶液中添加氯甲酸4-硝基苯酯39 (524 mg, 2.6 mmol )和吡啶(210 µl, 2.6 mmol)。在1 h之后,将温度升至室温且添加甲胺(1.950 mL,3.9 mmol,2 M,THF中)。在10 min之后,蒸发溶剂且使残余物通过层析柱以得到化合物40a和40b的混合物(分别是乙酯和甲酯;420 mg,来自HPLC的40a/40b比率为3:1,两个步骤产率的为约53%,[M+1]+:计算值:603.3、实验值:603.4 (对于40a而言);计算值为589.3、实验值为589.4 (对于40b而言))。
化合物41a和41b.将化合物40a和49b的混合物(420 mg,约0.68 mmol)溶于3 mL DCM中,向其中添加HCl (4.8 mmol,1.2 mL,4 N,二氧杂环己烷中)。在5℃下1 h之后,蒸发溶剂且化合物41a (乙酯)和41b (甲酯)的混合物(比率为约3:1)未经进一步纯化即用于下一步骤中。
化合物43a和43b. 在5℃下,将化合物41a和41b (400 mg,≈0.72 mmol)、化合物42 (购自Anichem,170 mg,0.721 mmol)和乙酸(0.041 mL, 0.721 mmol)的混合物混合于DCM中。添加三乙酰氧基硼氢化钠(306 mg, 1.44 mmol)。在30 min之后,将混合物溶于EtOAc中。在使用7% K2CO3水溶液和盐水洗涤之后,干燥有机相,过滤并蒸发以得到残余物。在柱层析纯化(MeOH: DCM,0-7%梯度)之后,获得化合物43a (乙酯)和43b (甲酯) (310 mg,大约0.42 mmol,产率大约为58.3%,43a:43b比率为约3:1,[M+1]+,计算值:738.4、实验值:738 (对于43a而言);计算值:724.4、实验值:724 (对于43b而言))。
化合物45a和45b.在室温下,将化合物43a和43b的混合物(310 mg,大约0.42 mmol)溶于5 mL DCM中。向该溶液中添加2,6-二-叔丁基吡啶(161 mg, 0.840 mmol)和化合物44 (根据Peltier等人,2006制备, 73.8 mg,0.420 mmol)的2 mL DCM溶液。在半小时之后,添加Et3N (58.6 µl, 0.420 mmol)。然后将混合物溶于EtOAc中,其使用10%柠檬酸水溶液、NaHCO3饱和水溶液和盐水洗涤。干燥有机相,过滤并蒸发成残余物。在柱层析纯化之后,获得作为粘性油状物的化合物45a和45b (294 mg,大约0.334 mmol,产率为80%,45a:45b比率为3:1,[M+1]+,计算值:877.5、实验值:877 (对于45a而言);计算值:863.5、实验值:863 (对于45b而言))。
化合物46a和46b.将化合物45a和45b的混合物(100 mg,大约0.114 mmol)添加至20 mL MeOH中的Pd/C (65 mg, 10%)的悬浮液中。添加HCl (28.5 µL,0.114 mmol,4 M,二氧杂环己烷中)。将烧瓶抽真空并使用H2再填充,重复该过程三次。在2 h之后,过滤悬浮液且蒸发溶剂以得到残余物。在5℃下,添加500 uL DMF中的化合物5a (19.59 mg, 0.137 mmol)、HATU (43.4 mg, 0.114 mmol)和DIPEA (49.8 µl, 0.285 mmol)的悬浮液。在悬浮液变得均匀之后,添加作为DMF (1 mL)溶液的上述残余物。添加更多DIPEA以将pH调节至约12。在10 min之后,将混合物溶于EtOAc中,其使用10%柠檬酸水溶液、NaHCO3饱和水溶液和盐水洗涤。干燥分离的有机相,过滤并蒸发。使所得残余物通过层析柱以得到化合物46a和46b的混合物(分别是乙酯和甲酯,80 mg,约0.082 mmol,产率为71.9%,46a:46b比率为3:1,[M+1]+,计算值:976.5、实验值:976.5 (对于46a而言);计算值:962.5、实验值:962.5 (对于46b而言))。
化合物47a和47b.在5℃下,将HCl (256 µl,1.024 mmol,4 M,二氧杂环己烷中)添加至化合物46a和46b (200 mg, 0.205 mmol)的2 mL MeOH溶液中。在1 h之后,蒸发溶液并在高真空中干燥过夜以得到粘性油状物。在室温下,将该粘性油状物、Fmoc保护的瓜氨酸18a (81 mg, 0.205 mmol)和DIPEA (179 µl, 1.024 mmol)溶于2 mL DMF中。添加丙基膦酸酐(T3P,178 µL,0.410 mmol,2.3 M,EtOAc中)。在1 h之后,将反应混合物溶于EtOAc中,其使用NaHCO3饱和水溶液和盐水洗涤。在分离之后,干燥并蒸发,使所得残余物通过层析柱(MeOH: DCM,0-10%梯度)以得到化合物47a和47b的混合物(分别是乙酯和甲酯,150 mg,大约0.119 mmol,产率为约58.3%,47a:47b比率为3:1,[M+1]+,计算值:1255.6、实验值:1255.6 (对于47a而言);计算值:1241.6、实验值:1241.6 (对于47b而言))。
化合物49. 在室温下,将化合物47a和47b的混合物(200 mg,约0.165 mmol)溶于5 mL DMF (具有5%哌啶)中。在30 min之后,将溶液蒸发至干燥。将所得残余物与Boc保护的缬氨酸N-羟基琥珀酰亚胺酯48 (61.9 mg, 0.198 mmol)、5 mL DMF和DIPEA (87 µL, 0.496 mmol)混合。在使反应进行过夜之后,将反应混合物蒸发至干燥。将所得混合物溶于5 mL MeOH、THF和水的混合物(1:1:1)中。添加NaOH且最终溶液的pH为14。在使反应在室温下进行过夜之后,使用HCl将混合物酸化至pH为3并在高真空下蒸发。使用TFA处理所获得固体且在10 min之后蒸发混合物,从而在制备型HPLC纯化之后提供化合物49 (80 mg,0.072 mmol,产率为43.8%,[M+1]+,计算值:1104.6、实验值:1104.6)。
化合物(III-4).将化合物49 (80 mg, 0.072 mmol)、市售化合物21a (Aldrich, 26.6 mg, 0.087 mmol)和DIPEA (38.0 µl, 0.217 mmol)溶于2 mL DMF中。在使反应进行过夜之后,蒸发混合物且通过制备型HPLC纯化残余物以得到主要异构体(15 mg,产率为16%,1/2[M+2]2+,计算值:649.3、实验值:649.5)和次要异构体(3.7 mg,产率为4%,1/2[M+2]2+,计算值:649.3、实验值:649.5))。主要异构体试验性地确定为在Tup亚单元的α-甲基处具有天然妥布赖森立体化学的(III-4),且次要异构体确定为其中具有倒转立体化学的化合物(III-5)。
实施例5 -化合物(I-5)、(I-6)和(I-7)
小部分上述来自使用HCl处理化合物46a和46b的粘性油状物不偶联至化合物18a,但反而将其溶于THF、MeOH和水(1:1:1)的混合物中。将反应混合物的pH调节至14。在使反应进行过夜之后,蒸发一半反应混合物并通过制备型层析纯化以得到化合物(I-5) (1 m;M+1, 876.6), (I-6) (1 mg;M+1, 862.5)和(I-7) (1 mg;M+1, 848.5)。
实施例6 -化合物(I-1)
用于合成化合物(I-1)的方案显示于图7中。
化合物51. 将HCl (6 N, 0.2 mL)添加至MeOH (1 mL)中的化合物50 (Cheng等人,2011;50 mg,0.198 mmol)和2,2-二甲氧基丙烷(0.244 mL, 1.982 mmol)的溶液中。在将反应混合物在室温下搅拌过夜之后,蒸发溶剂以提供52.8 mg化合物51。MS: (+) m/z 267.2 (M+1)。
化合物52. 将MeOH (1 mL)中的化合物51 (52.8 mg, 0.198 mmol)和碳载钯(10 wt%, 8 mg)的混合物在H2气氛下搅拌过夜。然后过滤掉催化剂并蒸发溶剂以提供46.9 mg化合物52。MS: (+) m/z 237.3 (M+1)。
化合物(I-1). 将DCM (0.5 mL)中的五氟苯酚(2.493 mg, 0.014 mmol)、1,3-二环己基碳化二亚胺(2.049 mg, 9.93 µmol)和化合物9 (5 mg, 9.03 µmol)的混合物在室温下搅拌过夜。然后蒸发溶剂。
向DMF (0.2 mL)中的所得残余物(6.50 mg, 9.03 µmol)和化合物52 (4.27 mg, 18.06 µmol)的溶液中添加DIEA (1滴)。在将反应混合物在室温下搅拌10 min之后,通过添加乙腈和含有0.1% TFA的水的1:1混合物(4 mL)来猝灭反应。制备型HPLC纯化提供2.5 mg作为白色固体的化合物(I-1)。MS: (+) m/z 772.5 (M+1)。
实施例6 -化合物(I-4)
图8显示用于合成化合物(I-4)的方案。
将DIEA添加至DMF (0.3 mL)中的化合物9 (10.69 mg, 0.019 mmol)和HATU (7.34 mg, 0.019 mmol)的溶液中,从而将pH调节至8-9。在将反应混合物在室温下搅拌10 min之后,添加DMF (0.5 mL)中的化合物53 (4 mg,0.019 mmol;根据Sani等人,2007制备)和DIEA,从而将pH调节至8-9。在将反应混合物在室温下搅拌20 min之后,通过添加乙腈和含有0.1% TFA的水的1:1混合物(4 mL)来猝灭反应。制备型HPLC纯化提供12.5 mg作为白色固体的化合物(I-4)。MS: (+) m/z 743.4 (M+1)。
实施例7 -硫代氨基甲酸酯
可通过使用氢化钠和然后硫代异氰酸酯处理合适前体(例如化合物6) (图1)来制备其中W是S的根据式(I)的化合物(即硫代氨基甲酸酯),如下所说明:
。
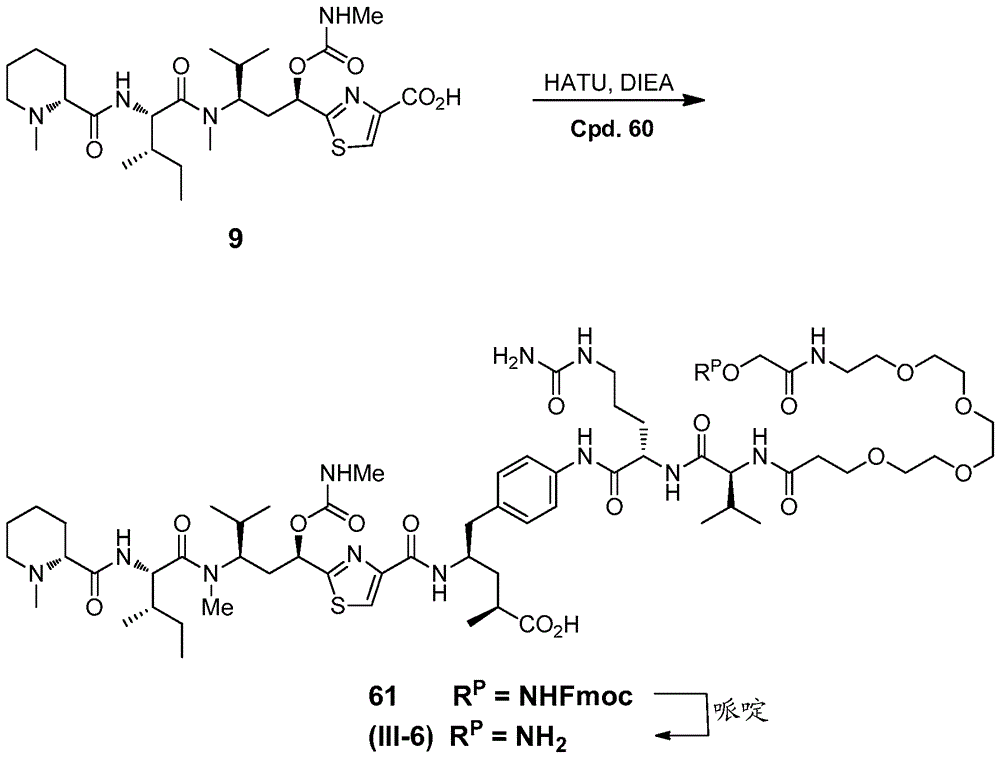
实施例8 -化合物(III-6)
用于合成化合物56的方案显示于图9a和9b中。
化合物56. 在室温下,将1,3-二环己基碳化二亚胺(DCC, 0.160 g, 0.778 mmol)添加至DCM (5 mL)中的1-氨基-3,6,9,12-四氧杂十五烷-15-酸叔丁酯54 (Quanta Biosciences, 0.25 g, 0.778 mmol)和2-(((((9H-茀-9-基)甲氧基)羰基)氨基)氧基)乙酸55 (Chem-Impex, 0.244 g, 0.778 mmol)的溶液中。在将反应混合物在室温下搅拌过夜之后,过滤掉沉淀物。然后浓缩滤液。通过快速层析(从使用DCM中的0-10%甲醇的梯度的硅胶洗脱)纯化粗产物以提供0.30 g作为无色油状物的化合物56。MS: (+) m/z 617.4 (M+1)。
化合物57. 将TFA (2 mL, 0.662 mmol)中的化合物56 (0.303 g, 0.491 mmol)的溶液在室温下搅拌2 h。在浓缩滤液之后,使用己烷洗涤残余物以提供0.28 g化合物57。MS: (+) m/z 561.3 (M+1)。
化合物58. 在室温下,将DCC (0.199 g, 0.963 mmol)添加至DCM (5 mL)中的化合物57 (0.27 g, 0.482 mmol)和1-羟基吡咯烷-2,5-二酮(也称为N-羟基琥珀酰亚胺或NHS,0.111 g,0.963 mmol)的溶液中。在将反应混合物在室温下搅拌过夜之后,过滤掉固体。然后浓缩滤液。通过快速层析(从使用己烷中的0-100%乙酸乙酯的梯度的硅胶洗脱)纯化粗产物以提供0.12 g作为无色油状物的化合物58。MS: (+) m/z 658.3 (M+1)。
化合物59. 在室温下,将DIEA (2滴)添加至DMF (2 mL)中的化合物58 (0.12 g, 0.182 mmol)和化合物21 (0.106 g, 0.182 mmol)的溶液中。在将反应混合物在室温下搅拌1 h之后,通过添加乙腈和含有0.1% TFA的水的混合物来猝灭反应。通过制备型HPLC纯化粗产物以提供0.12 g作为白色固体的化合物59。MS: (+) m/z 1121.6 (M+1)。
化合物60. 在室温下,将TFA (0.5 mL)添加至DCM (1 mL)中的化合物59 (20 mg, 0.018 mmol)的溶液中。在将反应混合物在室温下搅拌20 min之后,浓缩滤液以提供18.2 mg化合物60。MS: (+) m/z 1021.6 (M+1)。
化合物61. 将DIEA添加至DMF (0.4 mL)中的化合物9 (9.87 mg, 0.018 mmol)和HATU (6.78 mg, 0.018 mmol)的溶液中。将反应混合物的pH调节至8-9。在将反应混合物在室温下搅拌10 min之后,添加DMF (1 mL)中的化合物60 (18.2 mg, 0.018 mmol)和DIEA。将反应混合物的pH调节至8-9。在将反应混合物在室温下搅拌10 min之后,通过添加10 mL含有0.1% TFA的水和乙腈的1:1 (v/v)混合物来猝灭反应。通过制备型HPLC纯化粗产物以提供25 mg作为白色固体的化合物61。MS: (+) m/z 779.0 (M/2+1)。
化合物(III-6). 在室温下,将一滴哌啶添加至DMF (1 mL)中的化合物61 (25 mg, 0.016 mmol)的溶液中。在将反应混合物在室温下搅拌1 h之后,通过添加乙腈和含有0.1% TFA的水的混合物来猝灭反应。通过制备型HPLC纯化粗产物以提供20 mg作为白色固体的化合物(III-6)。MS: (+) m/z 668.0 (M/2+1)。化合物(III-6)具有羟胺基团,其可用于经由肟形成(例如)与已引入蛋白中的对乙酰基苯丙氨酸残基的酮基团进行缀合,如上文讨论。
使用3H胸苷掺入测定,化合物(III-6)和经修饰仪含有对乙酰基苯丙氨酸残基的抗间皮素抗体的缀合物针对N87胃癌细胞展现0.14的EC50。
在一个实施方案中,本发明提供化合物(III-6)和经修饰以含有酮基团的抗间皮素抗体的缀合物。优选地,修饰通过将对乙酰基苯丙氨酸残基并入抗体多肽链中来实现。还优选地,如此修饰的抗体是6A4。
实施例9 -中间体化合物69
图10显示用于合成化合物69的方案,该化合物可用作用于合成本发明化合物的中间体。
化合物63. 在5℃下,将化合物62 (Chem-Impex, 5 g, 23.8 mmol)添加至20 mL MeOH中的SOCl2 (3.47 mL, 47.6 mmol)的溶液中。在使反应进行过夜之后,将反应混合物在45℃下加热半小时。蒸发挥发性材料且将残余物溶于20 mL DCM中。添加Boc2O (7.8 g, 35.7 mmol)。使用Et3N将溶液pH调节至9 (使用湿润pH纸测试)。在数小时之后,将反应混合物溶于EtOAc中。使用10%柠檬酸溶液、NaHCO3饱和水溶液和盐水洗涤EtOAc溶液。干燥有机相,分离并蒸发。使来自蒸发干燥有机相的最终残余物通过柱以得到化合物63 (5 g,产率为65%,[M+1-Boc]+,计算值:225.1、实验值:225.2)。
化合物65. 在-78℃下,向20 mL化合物63 (2 g, 6.2 mmol)的DCM溶液中添加DIBAL-H (12.4 mL,12.4 mmol,1 M,DCM中)。在半小时之后,使用MeOH猝灭反应。使用HCl将溶液pH调节至约2。将混合物溶于EtOAc中,其使用10%柠檬酸、盐水洗涤,干燥,分离并蒸发。将残余物溶于30 mL DCM中。在0℃下,添加化合物64 (2.4g, 6.2 mmol, US 4,894,386)。在1 h之后于室温下蒸发混合物,使残余物通过柱以得到化合物65 (2 g,产率为79%,[M+1-Boc]+,计算值:307.2、实验值:307.1)。
化合物66. 在室温下,将化合物65 (600 mg, 1.48 mmol)溶于75 mL EtOAc中。将溶液转移至填充有N2和Pd/C (600 mg, 10%)的烧瓶中。将烧瓶抽真空并使用H2再填充;将循环重复三次。在1 h之后,过滤溶液并蒸发以得到作为差向异构体混合物的化合物66 (560 mg,产率为100%,[M+1]+,计算值:379.3、实验值:379.3)。直到后来再分离该混合物。
化合物67. 将化合物66 (1.30 g, 3.43 mmol)、Fmoc保护的瓜氨酸18a (1.6 g, 4.12 mmol)和2-乙氧基-1-乙氧基羰基-1,2-二氢喹啉(EEDQ, 1.06 g, 4.3 mmol)溶于DCM和MeOH的混合物(22 mL, 10:1)中。在使反应进行过夜之后,蒸发混合物且使残余物通过柱(MeOH:DCM,0-5%梯度)以得到作为差向异构体混合物(≈4:1,来自HPLC分析)的化合物67 ([M+1]+,计算值:758.4、实验值:758.4)。其未经进一步纯化即用于下一步骤中。
化合物68a. 在室温下,将来自上述反应的化合物67混合物溶于10 mL DMF (具有5%哌啶)中。在30 min之后,蒸发混合物并使用高真空泵干燥过夜。将残余物与化合物48 (1.5 g, 3.45 mmol)混合于5 mL DMF中。使用Et3N将溶液pH调节至12。在1 h之后,将反应混合物溶于EtOAc中,其使用10%柠檬酸、NaHCO3饱和水溶液和盐水洗涤。干燥有机相,过滤并蒸发至干燥。以与化合物67的去保护相同的方式使用DMF中的5%哌啶对残余物进行去保护。将在该步骤中获得的胺和化合物21a (1 g, 3.27 mmol)混合于10 mL DMF中。在室温下,使用Et3N将溶液pH调节至12。在使反应进行过夜之后,将混合物溶于EtOAc中,其使用10%柠檬酸、NaHCO3饱和水溶液和盐水洗涤。干燥有机相,过滤并蒸发以得到残余物。使残余物通过规则二氧化硅柱以得到化合物68a和68b的混合物(1g,来自化合物66的产率为35%,[M+1]+,计算值:828.5、实验值:828.5)。在制备型HPLC柱上分离之后,获得600 mg主要差向异构体(其结构试验性地确定为化合物68a,且化合物68b确定为次要差向异构体)。
化合物69. 将化合物68a (600 mg, 0.72 mmol)溶于5 mL DCM和TFA的混合物(1:1)中。在2 h之后,蒸发反应混合物以得到化合物69 (定量产率,[M+1],计算值:672.4、实验值:672.4)。
实施例10 -中间体化合物75
图11显示用于合成可用于合成本发明化合物的中间体化合物75的方案。
化合物71. 在室温下混合化合物70 (Cheng等人,2011,25 mg,0.044 mmol)、N,N'-二异丙基碳化二亚胺(DIC, 0.014 mL, 0.088 mmol)、4-(二甲基氨基)吡啶(DMAP, 10.78 mg, 0.088 mmol)和MeOH (0.036 mL, 0.882 mmol)。在一小时之后,蒸发反应混合物并通过柱以得到化合物71 (10 mg,产率为39%,M+1, 581.4)。
化合物72. 在5℃下,将化合物71 (122 mg,0.210 mmol,来自另一合成批次)溶于MeOH (2 mL)中。添加NaOMe (0.441 mL, 0.221 mmol)。在0.5 h之后,使用HCl (4 M,二氧杂环己烷中)中和混合物并蒸发以得到化合物72 (m+1, 539.4),其未经进一步纯化即用于下一步骤中。
化合物73. 在5℃下,将化合物72 (60 mg, 0.111 mmol)溶于DCM (1.5 mL)中。吡啶(0.045 mL, 0.557 mmol)。缓慢添加0.5 mL DCM中的氯甲酸4-硝基苯酯72a (67.3 mg, 0.334 mmol, Aldrich)。在使反应进行过夜之后,蒸发混合物并通过柱层析纯化以得到化合物73 (42 mg,0.060 mmol,产率为53.6%) (m+1, 704.4)。
化合物74. 在5℃下,将化合物73 (42 mg, 0.060 mmol)溶于DCM (1 mL)中。添加甲胺(1.853 mg, 0.060 mmol)。在0.5 h之后,蒸发混合物且使残余物通过层析柱(MeOH:DCM,0-15%梯度,产物在7-10%下洗脱出)以得到化合物74 (35 mg,0.059 mmol,产率为98%) (m+1, 596.4)。
化合物75. 在5℃下,将化合物74 (93 mg, 0.156 mmol)溶于THF (1 mL)中。添加0.34 mL水中的LiOH (7.47 mg, 0.94 mmol)。在反应完成之后,使用HCl (4 M,二氧杂环己烷中)中和反应混合物并在高真空中干燥以得到化合物75 (m+1, 582.3),其未经进一步纯化即用于下一步骤中。
实施例11 -化合物(III-7)和(III-8)
图12显示用于合成化合物(III-7)和(III-8)的方案。
化合物(III-7). 在DMF (0.5 mL)中使用HATU (6.83 mg, 0.018 mmol)和DIEA (14.86 µl, 0.085 mmol)活化化合物75 (11 mg, 0.019 mmol)。然后添加化合物69 (15.24 mg, 0.023 mmol)。在10 min之后,将反应混合物溶于DMSO中并通过制备型层析纯化以得到化合物(III-7) (12 mg,9.71 µmol,产率为51.4%) (m+1, 1235.7)。
化合物(III-8). 在DMF (0.5 mL)中使用HATU (6.21 mg, 0.016 mmol)和DIEA (0.014 mL, 0.077 mmol)活化化合物75 (10 mg, 0.017 mmol)。然后添加化合物23 (13.86 mg, 0.021 mmol)。在10 min之后,将反应混合物溶于DMSO中并通过制备型层析纯化以得到化合物(III-8) (13 mg,10.52 µmol,产率为61.2%) (m+1, 1235.7)。
实施例12 -化合物(I-8)和(I-9)
原则上,可通过使用蛋白酶组织蛋白酶B (二肽Val-Cit是其底物基序)分别处理化合物(III-7)和(III-8)来获得化合物(I-8)和(I-9)。具有邻位氨基的化合物(III-7)可对于裂解具有较大立体位阻。
实施例13 -化合物的生物活性
图13a和13b显示使用ATP发光测定的本发明的化合物(I-4)和(I-2)分别针对H226肺癌和786-O肾癌细胞的生物活性。作为对照,使用多柔比星和妥布赖森类似物化合物A (其在Tuv亚单元处含有乙酸酯基团代替氨基甲酸酯基团)。可根据Cheng等人,2011的教导来制备化合物A。
针对H226细胞,EC50值为:多柔比星,115.4 nM;化合物A,2.4 nM;和化合物(I-4),12.1 nM。针对786-O细胞,EC50值为:多柔比星,68.9 nM;化合物A,1.2 nM;和化合物(I-4),7.1 nM。
图13c和13d呈现类似类型的关于化合物(I-5)、(I-6)和(I-7)的数据。比较化合物是多柔比星和妥布赖森D。针对H226细胞的EC50值为:多柔比星,115.4 nM;妥布赖森D,0.05 nM;化合物(I-5),19.5 nM;化合物(I-6),9.9 nM;和化合物(I-7),15.4 nM。针对786-O细胞,EC50值为:多柔比星,68.9 nM;妥布赖森D,0.02 nM;化合物(I-5),22.9 nM;化合物(I-6),22.8 nM;和化合物(I-7),12.5 nM。
肿瘤细胞系从美国典型培养物保藏中心(American Type Culture Collection) (ATCC), P.O. Box 1549, Manassas, VA 20108, USA获得,且根据ATCC说明书培养。将细胞以1.0 × 103个细胞/孔接种于96孔板中保持3 h,用于ATP测定。将化合物的1:3连续稀释液添加至孔中。将板孵育24 h至72 h。使用CELLTITER-GLO®发光细胞活力试剂盒遵循制造商手册测量ATP板中的ATP水平,并在GLOMAX® 20/20发光计(都来自Promega, Madison,WI, USA)上读数。使用4.0版PRISM™软件(GraphPad Software, La Jolla, CA, USA)测定EC50值-药剂将细胞增殖抑制或减少50%的浓度。
实施例14 -缀合物的体外活性
图14显示缀合物(II-1)针对N87胃癌细胞(美国典型培养物保藏中心(ATCC), P.O. Box 1549, Manassas, VA 20108, USA)的体外活性。
将细胞分别以1.0 × 104个细胞/孔接种于96孔板中保持3 h,用于3H胸苷测定。将缀合物(II-1)的连续稀释液(1:3)添加至所述孔中。将板孵育120 h。在总孵育时段的最后24 h内用每孔1.0 μCi 3H-胸苷对板进行脉冲,收获,且在Top Count闪烁计数器(Packard Instruments, Meriden, CT)上读数。使用4.0版PRISM™软件(GraphPad Software, La Jolla, CA, USA)测定EC50值(药剂将细胞增殖抑制或减少最大抑制的50%的浓度)为0.2 nM。
来自图13a-13d和图14的EC50值的比较说明了两点。首先是与以下有关的效力增强:经由缀合物靶向递送细胞毒素,且然后通过使缀合物的抗体组分结合至其抗原来触发有效内在化机制(Schrama等人,2006)。第二,未缀合毒素是相对极性化合物且在不存在活性内在化机制下难以扩散穿过细胞膜,因此导致所测量EC50值较高(较低效力)。
实施例15 -缀合物的体内活性
在本实施例中,比较缀合物(II-1)与缀合物B的体内活性(Cheng等人,2011),缀合物B在结构上与缀合物(II-1)相同,除了其在Tuv亚单元中具有乙酸酯代替氨基甲酸酯:
。
将再悬浮于0.1 mL磷酸盐缓冲盐水(“PBS”)与0.1 mL基质胶(matrigel)中的5百万个OVCAR3卵巢癌细胞经皮下植入SCID小鼠的侧面区域。28天后开始肿瘤测量,且将小鼠随机化成7只小鼠的组,其中每一组的平均肿瘤大小通过肿瘤的LWH/2估计为60 mm3。在肿瘤植入后29天,腹膜内单次向小鼠给予测试化合物。图15显示,针对OVCAR3异种移植物,缀合物(II-1)比缀合物B更有效地抑制肿瘤生长。20天之后,差异尤其明显。
图16a、16b、17a、17b、18a、18b、19a、19b和20呈现本发明缀合物的其它体内效价数据,其根据上述方案(加以必要的变通)生成。
图16a显示对于一系列化合物(III-1)与抗CD70抗体1F4或抗间皮素抗体6A4的缀合物的OVCAR3 (卵巢癌)肿瘤体积对时间的数据。图注依次提供DAR (例如“2.7”)和剂量(µmol/kg,例如“0.1”)。在每一情况下,施用模式以单一剂量(SD)经腹膜内施用,除了最后的数据组(♦),其中以Q7D × 3施用缀合物。图16b显示相同实验的体重变化。
人单克隆抗体6A4的制备和表征描述于Terrett等人,2012中,其公开内容通过引用并入本文。抗体6A4的VH CDR1、CDR2和CDR3以及VK CDR1、CDR2和CDR3序列分别以SEQ ID NO:l、SEQ ID NO:2、SEQ ID NO:3、SEQ ID NO:4、SEQ ID NO:5和SEQ ID NO:6给出。抗体6A4的可变区VH和VK序列分别以SEQ ID NO:7和SEQ ID NO:8提供。
人单克隆抗体1F4的制备和表征描述于Coccia等人,2010中,其公开内容通过引用并入本文。抗体1F4的VH CDR1、CDR2和CDR3以及VK CDR1、CDR2和CDR3序列分别以SEQ ID NO:9、SEQ ID NO:10、SEQ ID NO:11、SEQ ID NO:12、SEQ ID NO:13和SEQ ID NO:14给出。抗体1F4的可变区VH和VK序列分别以SEQ ID NO:15和SEQ ID NO:16提供。
图17a和17b呈现类似实验的结果,但使用化合物(III-8)和抗间皮素抗体6A4的缀合物。在每一情况下,腹膜内施用单一剂量。
化合物(III-1)和抗间皮素抗体6A4的缀合物针对H226 (肺癌)肿瘤的功效呈现于图18a和18b中,其中DAR和剂量信息同样参见图注。在每一情况下,以单一剂量腹膜内施用缀合物。
图19a和19b呈现关于化合物(III-8)和抗间皮素抗体6A4的缀合物的H226数据。在第一数据组(●)中,施用为单一剂量,而对于最后两个数据组(▼和♦),施用方案为Q7D × 3。
图20显示化合物(III-1)与抗间皮素抗体6A4或抗CD70抗体1F4的缀合物针对N87胃癌肿瘤的功效数据。
抗体(尤其在用于缀合物中时)可具有天然恒定区或经设计以减小或消除效应子功能(例如ADCC)的工程改造(修饰)恒定区。此类修饰抗体恒定区的实施例由SEQ ID NO:25和SEQ ID NO:26的多肽提供。SEQ ID NO:25是用某些氨基酸取代修饰的IgG4同种型的恒定区。SEQ ID NO:26是杂合IgG1/IgG4恒定区。在SEQ ID NO:25和SEQ ID NO:26两者中,C-末端赖氨酸的存在是任选的。
实施例16 -稳定性研究
在本实施例中,比较具有各自氨基甲酸酯和乙酸酯基团的缀合物(II-1)和缀合物B的小鼠血清稳定性。
以0.1 µmol/kg的剂量将缀合物注射至小鼠中。在缀合物(II-1)的情况下,施用浓度为3.4 mg/mL且缀合物具有4.2的替换比(SR)。在缀合物B的情况下,施用浓度为1.2 mg/mL且SR为2.3。在每一时间点从3只动物中的每一只获取大约100 uL血清用于分析。
合并来自不同动物的血清样品,从而在每一时间点得到200-300 uL。离心合并体积以去除固体且将上清液用于分析。通过免疫亲和力捕获使用偶联至SEPHAROSE™珠粒的抗独特型单克隆抗体从血清分离缀合物。在捕获之后,通过暴露于低pH随后使用Tris碱中和来洗脱缀合物。通过添加活化组织蛋白酶B以裂解Cit-Val肽接头来释放存在于缀合物上的细胞毒素。在37℃下进行组织蛋白酶B消化3 h,随后添加1体积冷甲醇。通过LC-MS使用在线ESI-TOF MS与UPLC利用反相层析(Acquity HSS T3 2.1 × 50 mm)分析溶剂萃取的细胞毒素。
对于缀合物(II-1),在直至240 h的所有时间点中的任一时间,未检测到来自氨基甲酸酯基团水解的羟基化合物的存在。仅检测到氨基甲酸酯化合物。
相反,对于缀合物B,可在6小时之后检测到水解产物且直至72 h已发生约50%水解。下表2基于各个双价带电离子的质谱强度显示乙酸根和羟基化合物的相对量(M[H2+]372.2 Da和351.2 Da):
这些结果显示,尽管在Tuv亚单元中代替乙酸酯基团导致稳定得多的化合物,然而其仍保留实质性生物活性。
实施例17 –化合物72a、72b和72c
本实施例描述在氨基甲酸酯基团处具有结构变化的本发明化合物的制备和性质。参照图21的合成方案。
化合物70a. 将MeOH中的氨(2M, 0.089 mL, 0.178 mmol)添加至MeOH (2 mL)中的化合物7 (0.1 g, 0.148 mmol)中。在将反应混合物在室温下搅拌20 min之后,LCMS显示反应完成。蒸发溶剂。通过快速层析(从使用DCM中的0-15% MeOH的梯度的硅胶洗脱)纯化粗产物以提供55 mg作为白色固体的化合物70a。MS: (+) m/z 554.3 (M+1)。
化合物71a 在室温下,将水(0.5 mL)中的LiOH (4.41 mg, 0.184 mmol)添加至THF (1 mL)中的化合物70a (51 mg, 0.092 mmol)的溶液中。在将反应混合物在室温下搅拌2 h之后,LCMS显示反应完成。蒸发溶剂。通过快速层析(从使用DCM中的0-30% MeOH的梯度的硅胶洗脱)纯化粗产物以提供47 mg作为白色固体的化合物71a。MS: (+) m/z 540.3 (M+1)。
化合物72a. 将DIEA (6.45 µL, 0.037 mmol)添加至DMF (0.5 mL)中的化合物71a (20 mg, 0.037 mmol)和HATU (14.09 mg, 0.037 mmol)的溶液中。将反应混合物的pH调节至8-9。在将反应混合物在室温下搅拌20 min之后,添加DMF (1 mL)中的化合物23 (24.9 mg, 0.037 mmol)和DIEA (6.45 µL, 0.037 mmol)。将反应溶液的pH调节至8-9。在将反应混合物在室温下再搅拌20 min之后,LCMS显示反应完成。通过添加10 mL水(含有0.1% TFA)和乙腈(含有0.1% TFA)的1:1 (v/v)混合物来猝灭反应。通过制备型HPLC纯化粗产物以提供35.2 mg作为白色固体的化合物72a。MS: (+) m/z 1193.6 (M+1)。化合物72a也在上文中称为化合物(III-9)。
化合物72a和抗CD70抗体1F4的缀合物针对786-O肾癌细胞具有0.19 nM的EC50。化合物72a和抗间皮素抗体6A4的缀合物针对N87胃癌细胞具有0.16 nM的EC50。在两种情况下,使用3H-胸苷掺入测定。
化合物70b. 将DMF (1.8 mL)中的化合物7 (0.1 g, 0.148 mmol)和苯胺(0.041 mL, 0.444 mmol)的溶液在50℃下加热过夜。蒸发溶剂。通过快速层析(从使用DCM中的0-15% MeOH的梯度的硅胶洗脱)纯化粗产物以提供58 mg作为白色固体的化合物5。MS: (+) m/z 630.4 (M+1)。
化合物71b. 在室温下,将水(0.5 mL)中的LiOH (4.03 mg, 0.168 mmol)添加至THF (1 mL)中的化合物70b (53 mg, 0.084 mmol)的溶液中。在将反应混合物在室温下搅拌2 h之后,LCMS显示反应完成。蒸发溶剂。通过快速层析(从使用DCM中的0-30% MeOH的梯度的硅胶洗脱)纯化粗产物以提供43 mg作为白色固体的化合物71b。MS: (+) m/z 616.3 (M+1)。
化合物72b. 将DIEA (5.66 µL, 0.032 mmol)添加至DMF (0.5 mL)中的化合物71b (20 mg, 0.032 mmol)和HATU (12.35 mg, 0.032 mmol)的溶液中。将反应混合物的pH调节至8-9。在将反应混合物在室温下搅拌20 min之后,添加DMF (1 mL)中的化合物23 (21.82 mg, 0.032 mmol)和DIEA (5.66 µL, 0.032 mmol)。将反应溶液的pH调节至8-9。在将反应混合物在室温下再搅拌20 min之后,LCMS显示反应完成。通过添加10 mL水(含有0.1% TFA)和乙腈(含有0.1% TFA)的1:1 (v/v)混合物来猝灭反应。通过制备型HPLC纯化粗产物以提供35 mg作为白色固体的化合物72b。MS: (+) m/z 1269.7 (M+1)。化合物72b也在上文中称为化合物(III-10)。
化合物72b和抗CD70抗体1F4的缀合物针对786-O肾癌细胞具有0.58 nM的EC50。化合物72b和抗间皮素抗体6A4的缀合物针对N87胃癌细胞具有0.47 nM的EC50。在两种情况下,使用3H胸苷掺入测定。
化合物70c. 在室温下,将DMF中的二甲胺(1 mL, 0.148 mmol)逐滴添加至DMF (1 mL)中的化合物7 (0.1 g, 0.148 mmol)的溶液中,直至反应完成。蒸发溶剂。通过快速层析(从使用DCM中的0-15% MeOH的梯度的硅胶洗脱)纯化粗产物以提供56 mg作为白色固体的化合物70c。MS: (+) m/z 582.3 (M+1)。
化合物71c. 在室温下,将水(0.5 mL)中的LiOH (8.23 mg, 0.344 mmol)添加至THF (1 mL)中的化合物70c (0.1 g, 0.172 mmol)的溶液中。在将反应混合物在室温下搅拌2 h之后,LCMS显示反应完成。蒸发溶剂。通过快速层析(从使用DCM中的0-30% MeOH的梯度的硅胶洗脱)纯化粗产物以提供50.8 mg作为白色固体的化合物71c。MS: (+) m/z 568.3 (M+1)。
化合物72c. 将DIEA (6.14 µL, 0.035 mmol)添加至DMF (0.5 mL)中的化合物71c (20 mg, 0.035 mmol)和HATU (13.39 mg, 0.035 mmol)的溶液中。将反应混合物的pH调节至8-9。在将反应混合物在室温下搅拌20 min之后,添加DMF (1 mL)中的化合物23 (23.67 mg, 0.035 mmol)和DIEA (6.14 µL, 0.035 mmol)。将反应溶液的pH调节至8-9。在将反应混合物在室温下再搅拌20 min之后,LCMS显示反应完成。通过添加10 mL水(含有0.1% TFA)和乙腈(含有0.1% TFA)的1:1 (v/v)混合物来猝灭反应。通过制备型HPLC纯化粗产物以提供23.8 mg作为白色固体的化合物72c。MS: (+) m/z 1221.6 (M+1)。化合物72c也在上文中称为化合物(III-11)。
化合物72c和抗CD70抗体1F4的缀合物针对786-O肾癌细胞具有0.32 nM的EC50。化合物72c和抗间皮素抗体6A4的缀合物针对N87胃癌细胞具有0.34 nM的EC50。在两种情况下,使用3H胸苷掺入测定。
实施例18 -化合物75a、75b和75c
本实施例描述先前实施例的妥布赖森类似物(但不连接接头部分)的制备。参照图22的合成方案。
化合物73. 在室温下,将水(5 mL)中的LiOH (0.071 g, 2.97 mmol)添加至THF (5 mL)中化合物18 (0.25 g, 0.743 mmol)的溶液中。在将反应混合物在室温下搅拌2 h之后,蒸发溶剂。通过快速层析(从使用DCM中的0-20% MeOH的梯度的硅胶洗脱)纯化粗产物以提供0.22 g作为白色固体的化合物73。MS: (+) m/z 223.3 (M+1-Boc)。
化合物74. 将1,4-二氧杂环己烷(4 mL, 0.620 mmol)中的化合物73 (0.2 g, 0.620 mmol)和4 N HCl的混合物在室温下搅拌1 h。蒸发溶剂。使用己烷将白色固体化合物74洗涤两次。MS: (+) m/z 223.3 (M+1)。
化合物75a. 将DIEA (3.23 µL, 0.019 mmol)添加至DMF (0.5 mL)中的化合物71a (10 mg, 0.019 mmol)和HATU (7.05 mg, 0.019 mmol)的溶液中,从而将pH调节至8-9。在将反应混合物在室温下搅拌20 min之后,添加DMF (0.5 mL)中的DIEA (3.23 µL, 0.019 mmol)和化合物74 (5.35 mg, 0.024 mmol),从而将pH调节至8-9。在将反应混合物在室温下搅拌1 h之后,LCMS显示反应完成。通过添加10 mL乙腈和含有0.1% TFA的水的1:1 (v/v)混合物来猝灭反应。通过制备型HPLC纯化粗产物以提供6.0 mg作为白色固体的化合物75a。MS: (+) m/z 744.4 (M+1)。化合物75a也在上文中称为化合物(I-10)。
使用ATP发光测定,化合物75a针对N87胃癌细胞具有75.8 nM的EC50。
化合物75b. 将DIEA (2.83 µL, 0.016 mmol)添加至DMF (0.5 mL)中的化合物71b (10 mg, 0.016 mmol)和HATU (6.17 mg, 0.016 mmol)的溶液中,从而将pH调节至8-9。在将反应混合物在室温下搅拌20 min之后,添加DMF (0.5 mL)中的DIEA (2.83 µL, 0.016 mmol)和化合物74 (4.69 mg, 0.021 mmol),从而将pH调节至8-9。在将反应混合物在室温下搅拌1 h之后,LCMS显示反应完成。通过添加10 mL乙腈和含有0.1% TFA的水的1:1 (v/v)混合物来猝灭反应。通过制备型HPLC纯化粗产物以提供7.6 mg作为白色固体的化合物75b。MS: (+) m/z 820.4 (M+1)。化合物75b也在上文中称为化合物(I-11)。
使用ATP发光测定,化合物75b针对N87胃癌细胞具有0.39 nM的EC50。
化合物75c. 将DIEA (3.07 µL, 0.018 mmol)添加至DMF (0.5 mL)中的化合物71c (10 mg, 0.018 mmol)和HATU (6.70 mg, 0.018 mmol)的溶液中,从而将pH调节至8-9。在将反应混合物在室温下搅拌20 min之后,添加DMF (0.5 mL)中的DIEA (3.07 µL, 0.018 mmol)和化合物74 (5.09 mg, 0.023 mmol),从而将pH调节至8-9。在将反应混合物在室温下搅拌1 h之后,LCMS显示反应完成。通过添加10 mL乙腈和含有0.1% TFA的水的1:1 (v/v)混合物来猝灭反应。通过制备型HPLC纯化粗产物以提供7.6 mg作为白色固体的化合物75c。MS: (+) m/z 772.5 (M+1)。化合物75c也在上文中称为化合物(I-12)。
使用ATP发光测定,化合物75c针对N87胃癌细胞具有5.4 nM的EC50。
实施例19–化合物80
本实施例描述适于经由“点击”化学缀合的化合物80的合成,该化合物具有可与配偶体分子的叠氮化物基团发生反应的环辛炔基团。相应合成方案显示于图23中。
化合物77. 在室温下,将DCC (22.40 mg, 0.109 mmol)添加至DCM (1 mL)中的DBCO-PEG4-酸76 (50 mg,0.090 mmol,购自Click Chemistry Tools, Scottsdale, AZ)和N-羟基琥珀酰亚胺(NHS, 20.83 mg, 0.181 mmol)的溶液中。将反应混合物在室温下搅拌过夜。过滤掉固体,且浓缩滤液以提供化合物77。MS: (+) m/z 650.3 (M+1)。
化合物78. 在室温下,将DIEA添加至DMF (1 mL)中的化合物77 (58.8 mg, 0.091 mmol)和化合物21 (57.6 mg, 0.100 mmol)的溶液中,从而将pH调节至8-9。在将反应混合物在室温下搅拌1 h之后,LCMS显示反应完成。蒸发溶剂。通过制备型HPLC纯化粗产物以提供20 mg作为白色固体的化合物78。MS: (+) m/z 1113.6 (M+1)。
化合物79. 在室温下,将TFA (0.5 mL, 6.53 mmol)添加至DCM (1 mL)中的化合物78 (17.2 mg, 0.015 mmol)的混合物中。在将反应混合物在室温下搅拌1 h之后,LCMS显示反应完成。蒸发溶剂以提供化合物79。MS: (+) m/z 1013.5 (M+1)。
化合物80. 将DIEA (2.96 µL, 0.017 mmol)添加至DMF (0.4 mL)中的化合物9 (8.55 mg, 0.015 mmol)和HATU (5.87 mg, 0.015 mmol)的溶液中。将反应混合物的pH调节至8-9。在将反应混合物在室温下搅拌20 min之后,添加DMF (1 mL)中的化合物79 (15.65 mg, 0.015 mmol)和DIEA (2.96 µL, 0.017 mmol)。将反应混合物的pH调节至8-9。在将反应混合物在室温下再搅拌20 min之后,LCMS显示反应完成。蒸发溶剂。将粗产物再溶于1 mL DMSO中,并通过制备型HPLC纯化以提供作为白色固体的化合物80。MS: (+) m/z 775.0 (M/2+1)。化合物80也在上文中称为化合物(III-12)。
使用3H胸苷掺入测定,化合物80和抗磷脂酰肌醇蛋白聚糖3抗体4A6的7种变体(经修饰以在各个位置处具有叠氮化物基团)的缀合物针对N87胃癌细胞展现1.4 nM、0.17 nM、0.13 nM、0.22 nM、0.14 nM、0.064 nM和0.25 nM的EC50。
抗体4A6的制备和表征描述于Terrette等人,2010中,其公开内容通过引用并入本文。抗体4A6的VH CDR1、CDR2和CDR3以及VK CDR1、CDR2和CDR3序列分别以SEQ ID NO:17、SEQ ID NO:18、SEQ ID NO:19、SEQ ID N提供。
在一个实施方案中,本发明提供化合物80和经修饰以包括叠氮化物基团的多肽、优选地抗体的缀合物。
实施例20–化合物88
本实施例描述具有可用于缀合的烷基伯胺官能团的化合物88的合成。参照图24的合成方案。
化合物82. 将DMF (3 mL)中的1-氨基-3,6,9,12-四氧杂十五烷-15-酸叔丁酯54 (0.285 g,0.888 mmol,购自VWR;还参见实施例8)和6-((((9H-茀-9-基)甲氧基)羰基)氨基)己酸2,5-二氧代吡咯烷-1-基酯81 (0.4 g,0.888 mmol,购自Chem-Impex)的混合物在室温下搅拌2 h。蒸发溶剂。通过快速层析(从使用DCM中的0-10% MeOH的梯度的硅胶洗脱)纯化粗产物以提供0.4 g作为白色固体的化合物82。MS: (+) m/z 657.4 (M+1)。
化合物83. 将TFA (2 mL, 0.393 mmol)中的化合物82 (0.258 g, 0.393 mmol)的混合物在室温下搅拌1 h。蒸发溶剂。使用己烷将化合物83 (白色固体)洗涤两次且未经进一步纯化即用于下一步骤反应中。
化合物84. 在室温下,将DCC (0.162 g, 0.786 mmol)添加至DCM (5 mL)中的化合物83 (0.236 g, 0.393 mmol)和NHS (0.090 g, 0.786 mmol)的混合物中。在将反应混合物在室温下搅拌过夜之后,过滤掉固体,并浓缩滤液。通过快速层析(从使用己烷中的0-100% EtOAc的梯度的硅胶洗脱)纯化粗产物以提供0.195 g作为无色油状物的化合物84。MS: (+) m/z 698.3 (M+1)。
化合物85. 在室温下,将DIEA添加至DMF (1.5 mL)中的化合物21 (0.162 g, 0.279 mmol)和化合物84 (0.195 g, 0.279 mmol)的溶液中。将反应混合物在室温下搅拌1 h之后,通过添加6 mL乙腈和含有0.1% TFA的水的1:1混合物来猝灭反应。通过制备型HPLC纯化粗产物以提供0.292 g作为白色固体的化合物85。MS: (+) m/z 1161.6 (M+1)。
化合物86. 在室温下,将TFA (0.5 mL)添加至DCM (1 mL)中的化合物85 (22.1 mg, 0.019 mmol)的混合物中。在将反应混合物在室温下搅拌20 min之后,蒸发溶剂以提供化合物86。MS: (+) m/z 1061.6 (M+1)。
化合物87. 将DIEA添加至DMF (0.4 mL)中的化合物9 (10.53 mg, 0.019 mmol)和HATU (7.23 mg, 0.019 mmol)的溶液中。将反应混合物的pH调节至8-9。在将反应混合物在室温下搅拌10 min之后,添加DMF (1 mL)中的化合物86 (20.19 mg, 0.019 mmol)和DIEA。将反应溶液的pH调节至8-9。在将反应混合物在室温下搅拌10 min之后,LCMS显示反应完成。通过添加10 mL水(0.1% TFA)和乙腈的1:1 (v/v)混合物来猝灭反应。通过制备型HPLC纯化粗产物以提供27.3 mg作为白色固体的化合物87。MS: (+) m/z 799.1 (M/2+1)。
化合物88. 在室温下,将哌啶添加至DMF (2 mL)中的化合物87 (27.3mg, 0.017 mmol)的溶液中,从而将pH调节至9-10。在将反应混合物在室温下搅拌1 h之后,通过添加6 mL乙腈和含有0.1% TFA的水的1:1混合物来猝灭反应。通过制备型HPLC纯化粗产物以提供22.5 mg作为白色固体的化合物88。MS: (+) m/z 688.1 (M/2+1)。化合物88也在上文中称为化合物(III-13)。
在一个实施方案中,提供缀合物,其中化合物88经由在化合物88的烷基伯胺与多肽中氨基酸(优选地谷氨酸)的侧链残基上的羧基之间形成酰胺缀合至多肽(其优选是抗体)。本发明还提供制备此类缀合物的方法,其包括在酶转谷氨酰胺酶存在下组合化合物88和多肽。
本发明的上文详述包括主要地或排他性地与本发明的特定部分或方面有关的段落。应理解,这是出于清晰和便利性,特定特征可不仅仅与公开其的段落相关,且本文中的公开内容包括见于不同段落中的信息的所有适当组合。类似地,尽管本文中的各个图和说明涉及本发明的具体实施方案,但应理解,在特定图或实施方案的背景中公开具体特征的情况下,也可在适当程度,在另一图或实施方案的背景下,与另一特征组合或通常在本发明中使用此类特征。
此外,尽管已在某些优选实施方案方面特定地描述本发明,但本发明不限于此类优选实施方案。而是,本发明范围由随附权利要求所定义。
参考文献
下面提供了本说明书前文中通过第一作者(或发明人)和日期的缩写方式引用的参考文献的全部引文。这些参考文献中的每一篇出于所有目的通过引用并入本文。下文中或本说明书中其它地方的参考文献的引用并非承认该参考文献是材料的现有技术。
Abe等人, WO 97/21712 (1997).
Boyd等人, US 2008/0279868 A1 (2008).
Boyd等人, US 7,691,962 B2 (2010).
Balasubramanian等人, Bioorg. Med. Chem. Lett. 2008, 18, 2996-2999.
Balasubramanian等人, J. Med. Chem. 2009, 52 (2), 238-240.
Chai等人, Chem. & Biol. 2010, 17(3), 296-309.
Chai等人, US 2011/0245295 A1 (2011).
Cheng等人, US 2011/0027274 A1 (2011).
Coccia等人, US 2010/0150950 (2010)
Davis等人, US 2008/0176958 A1 (2008).
Domling, DE 10 2004 030 227 A1 (2006).
Domling等人, US 2005/0239713 A1 (2005) [2005a].
Domling等人, US 2005/0249740 A1 (2005) [2005b].
Domling等人, Mol. Diversity 2005, 9, 141-147 [2005c].
Domling等人, Ang. Chem. Int. Ed. 2006, 45, 7235-7239.
Ellman等人, US 8,476,451 B2 (2013).
Hamel等人, Curr. Med. Chem. – Anti-Cancer Agents 2002, 2, 19-53.
Hoefle等人, DE 100 08 089 A1 (2001).
Hoefle等人, Pure Appl. Chem. 2003, 75 (2-3), 167-178.
Hoefle等人, US 2006/0128754 A1 (2006) [2006a].
Hoefle等人, US 2006/0217360 A1 (2006) [2006b].
Jackson等人, US 2013/0224228 A1 (2013).
Kaur等人, Biochem. J. 2006, 396, 235-242.
Khalil等人, ChemBioChem 2006, 7, 678-683.
Leamon等人, Cancer Res. 2008, 68 (23), 9839-9844.
Leamon等人, US 2010/0323973 A1 (2010).
Leung等人, US 2002/0169125 A1 (2002).
Low等人, US 2010/0324008 A1 (2010).
Lundquist等人, Org. Lett. 2001, 3, 781-783.
Neri等人, ChemMedChem 2006, 1, 175-180.
Pando等人, J. Am. Chem. Soc., 2011, 133, 7692-7696.
Patterson等人, Chem. Eur. J. 2007, 13, 9534-9541.
Patterson等人, J. Org. Chem. 2008, 73, 4362-4369.
Peltier等人, J. Am. Chem. Soc. 2006, 128, 16018-16019.
Reddy等人, Mol. Pharmaceutics 2009, 6 (5), 1518-1525.
Reichenbach等人 WO 98/13375 A1 (1998).
Richter, US 2012/0129779 A1 (2012) [2012a]
Richter, US 2012/0252738 A1 (2012) [2012b].
Richter, US 2012/0252739 A1 (2012) [2012c].
Sani等人, Angew. Chem. Int. Ed. 2007, 46, 3526-3529.
Sasse等人, J. Antibiotics 2000, 53 (9), 879-885.
Sasse等人, Nature Chem. Biol. 2007, 3 (2), 87-89.
Schluep等人, Clin. Cancer Res. 2009, 15 (1), 181-189.
Schrama等人, Nature Rev. Drug Disc. 2006, 5, 147-159.
Shankar等人, SYNLETT 2009, 8, 1341-1345.
Shankar等人, Org. Biomol. Chem., 2013, 11(14), 2273-2287.
Shibue等人, Tetrahedron Lett. 2009, 50, 3845-3848.
Shibue等人, Chemistry Eur. J., 2010, 16(38), 11678-11688.
Shibue等人, Bioorg. Med. Chem., 2011, 21, 431-434.
Sreejith等人, SYNLETT 2011, No. 12, 1673-1676x.
Steinmetz等人, Angew. Chem. Int. Ed. 2004, 43, 4888-4892.
Terrett等人, US 8,268,970 B2 (2012).
Terrette等人, US 2010/0209432 A1 (2010).
Ullrich等人, Angew. Chemie Int. Ed. 2009, 48, 4422-4425.
Vlahov等人, Bioorg. Med. Chem. Lett. 2008, 18 (16), 4558-4561 [2008a].
Vlahov等人, US 2008/0248052 A1 (2008) [2008b].
Vlahov等人, US 2010/0240701 A1 (2010) [2010a].
Vlahov等人, US 2010/0048490 A1 (2010) [2010b].
Wang等人, Chem. Biol. Drug. Des 2007, 70, 75-86.
Wipf等人, Org. Lett. 2004, 6 (22), 4057-4060.
Wipf等人, Org. Lett. 2007, 9 (8), 1605-1607.
Wipf等人, US 2010/0047841 A1 (2010).
Zanda等人, US 8,580,820 B2 (2013)。
序列表
命名为“SEQT_12026WOPCT.txt”的序列表以其整体通过引用并入本文,其包含SEQ ID NO:1至SEQ ID NO:26,其包括本文公开的核酸和/或氨基酸序列。序列表已以ASCII文本格式经由EFS-Web一同递交,且由此构成纸件与其计算机可读取形式二者。首先于2014年1月4日使用PatentIn 3.5产生序列表且大小为大约15 KB。
下表3概述本申请中公开的序列的说明。
- 还没有人留言评论。精彩留言会获得点赞!