用于治疗癌症的PD-l拮抗剂和RAF抑制剂的组合的制作方法
本申请公开了用于预防癌症、延缓癌症进展或治疗癌症的药物组合,其中所述药物组合展现出协同功效。所述药物组合包含针对pd-1的人源化拮抗剂单克隆抗体和raf抑制剂。本申请还公开了用于在受试者中预防癌症、延缓癌症进展或治疗癌症的方法,其包括向所述受试者施用治疗有效量的针对pd-1的人源化拮抗剂单克隆抗体和治疗有效量的raf抑制剂。
背景技术:
由ras-raf-mek-erk激酶级联构成的丝裂原活化蛋白激酶(mapk)信号传导途径是调节多种细胞活性诸如细胞存活、生长、分化和增殖的关键信号转导途径之一。通常在人类癌症中观察到导致mapk途径组成性活化的遗传畸变。已在多种人类恶性肿瘤中检测到致癌性b-raf突变,其中v600e突变占至少90%。选择性靶向于突变的b-raf的抑制剂诸如威罗菲尼(vemurafenib)和达拉菲尼(dabrafenib)已实现高响应率并被fda批准用于治疗携带b-rafv600e的黑素瘤患者。先前的研究表明b-raf靶向疗法可增加抗原表达,降低肿瘤微环境中的免疫抑制因子并改善t效应细胞向肿瘤的归巢。因此,选择性b-raf抑制剂被认为具有免疫增敏性质并可与免疫疗法联用以在治疗癌症中实现较持久的疾病控制/响应。
程序性死亡1蛋白(pd-1、pdcd-1或cd279)是一种与cd28/ctla4共刺激/抑制受体家族相关的55kd受体蛋白(blank等人,2005cancerimmunolimmunother54:307-314)。全长pd-1含有288个氨基酸残基(ncbi登录号:np_005009)。它的细胞外结构域由氨基酸残基1-167构成且胞质c末端尾部包含残基191-288,其具有两个假设的免疫调节基序即一个基于免疫受体酪氨酸的抑制基序(itim;vivier等人,1997immunoltoday18:286-291)和一个免疫受体酪氨酸切换基序(itsm;chemnitz等人,2004jimmunol173:945-954)。
已鉴定了两种与序列相关的配体即pd-l1(b7-h1)和pd-l2(b7-dc)与pd-1特异性相互作用,由此诱导细胞内信号转导,其抑制由cd3和cd28介导的t细胞活化(riley,2009immunolrev229:114-125),这又减弱t细胞活性,例如减少细胞增殖、il-2和ifn-γ分泌及其它生长因子和细胞因子分泌。
经常在诸如t细胞、b细胞、单核细胞和天然杀伤(nk)细胞的免疫细胞中发现pd-1的表达。其很少在诸如肌肉、上皮、神经元组织等的其它人类组织中表达。另外,高水平的pd-1表达常常与免疫细胞的活化相关。例如,当人t细胞系jurkat通过植物血凝素(pha)或佛波醇酯(12-o-十四酰基佛波醇-13-乙酸酯或tpa)来活化时,在western印迹中可见pd-1表达的上调(vibharka等人,1997expcellres232:25-28)。当通过抗cd3抗体进行刺激时,在经刺激的鼠类t和b淋巴细胞及原代人类cd4+t细胞中观察到相同的现象(agata等人,1996intimmunol8:765-772;bennett等人,2003jimmunol170:711-118)。pd-1表达在刺激t效应细胞后的增加再次使活化的t效应细胞趋向于耗竭和减少免疫活性。因此,由pd-1介导的抑制信号在免疫耐受中发挥重要作用(bour-jordan等人,2011immunolrev241:180-205)。
在涉及不同类型组织和器官的多种癌症中报告了pd-1表达在肿瘤浸润淋巴细胞(til)中的增加和pd-1配体表达在肿瘤细胞中的增加,所述组织和器官为诸如肺(konishi等人,2004clincancerres10:5094-5100)、肝(shi等人,2008intjcancer128:887-896;gao等人,2009clincancerres15:971-979)、胃(wu等人,2006actahistochem108:19-24)、肾(thompson等人,2004procnatlacadsci101:17174-17179;thompson等人,2007clincancerres13:1757-1761)、乳腺(ghebeh等人,2006neoplasia8:190-198)、卵巢(hamanishi等人2007procnatlacadsci104:3360-3365)、胰腺(nomi等人,2007clincancerres13:2151-2157)、黑素细胞(hino等人,2010cancer116:1757-1766)和食道(ohigashi等人,2005clincancerres11:2947-2953)。更经常地,pd-1和pd-l1表达在那些癌症中的增加与患者生存结果的不良预后是相关的。敲除pd-1基因由此抑制异体移植癌细胞生长的转基因小鼠进一步阐明了pd-1信号传导在用于癌症根治或耐受的免疫系统调节中的重要性(zhang等人,2009blood114:1545-1552)。
wo2013/097224a1公开了第二代b-raf抑制剂,其已表现出针对raf家族的丝氨酸/苏氨酸激酶的强效抑制活性。
pct申请pct/cn2016/079251在wo2013/097224a1中公开了第二代b-raf抑制剂的药用盐,具体为5-(((1r,1as,6br)-1-(6-(三氟甲基)-1h-苯并[d]咪唑-2-基)-1a,6b-二氢-1h-环丙并[b]苯并呋喃-5-基)氧基)-3,4-二氢-1,8-二氮杂萘-2(1h)-酮倍半马来酸盐(以下称为化合物1),其用于治疗在raf-mek-erkmapk途径中具有畸变(包括b-raf突变、k-ras/n-ras突变和nf1突变)的癌症,其具有针对raf家族激酶(包括a-raf、b-raf、c-raf和b-rafv600e)的强效和可逆抑制活性。
wo2015/035606a1公开了包含重链可变区(vh)和轻链可变区(vk)(分别包含seqidno24和seqidno26)及igg4重链效应子或恒定结构域(包含seqidno:88)的单克隆抗体(以下称为mab1),其特异性结合pd-1,尤其是包含k45和i93或i93、l95和p97的pd-1残基且在免疫细胞中抑制由pd-1介导的细胞信号传导和活性,结合至其配体结合所需要的一组氨基酸残基的抗体。
本申请发明人已发现上述抗pd1抗体(即mab1)及其抗体片段与选择性b-raf抑制剂(即化合物1)的组合令人惊讶且预料不到地在患有与k-ras突变相关的癌症的受试者中通过增加ifn-γ产生而增强t细胞响应。具体地,本申请发明人已预料不到地发现具体的抗pd1抗体(即mab1)和具体的选择性b-raf抑制剂(即化合物1)的组合与单独的所述抗pd1抗体或b-raf抑制剂的单一疗法相比在与k-ras突变相关的癌症中实现对肿瘤生长的协同抑制。
技术实现要素:
在第一个方面,本申请公开了用于预防癌症、延缓癌症进展或治疗癌症的药物组合,其包含针对pd-1的人源化拮抗剂单克隆抗体和raf抑制剂。所述药物组合在抑制癌症的肿瘤生长中产生协同功效。
在第二个方面,本申请公开了用于在受试者中预防癌症、延缓癌症进展或治疗癌症的方法,其包括向有此需要的受试者施用治疗有效量的针对pd-1的人源化拮抗剂单克隆抗体和治疗有效量的raf抑制剂。
在第三个方面,本申请公开了针对pd-1的人源化拮抗剂单克隆抗体,其用于与raf抑制剂组合以预防癌症、延缓癌症进展或治疗癌症。在该方面的一个实施方案中,本申请公开了raf抑制剂,其用于与针对pd-1的人源化拮抗剂单克隆抗体组合以预防癌症、延缓癌症进展或治疗癌症。
在第四个方面,本申请公开了药物组合在制备用于预防癌症、延缓癌症进展或治疗癌症的药物中的用途,所述药物组合包含针对pd-1的人源化拮抗剂单克隆抗体和raf抑制剂。
在第五个方面,本申请公开了制品或“试剂盒”,其包含第一容器、第二容器和包装插页,其中所述第一容器包含至少一个剂量的包含pd-1拮抗剂的药物,所述第二容器包含至少一个剂量的包含raf抑制剂的药物且所述包装插页包含使用所述药物用于治疗受试者癌症的说明。
附图说明
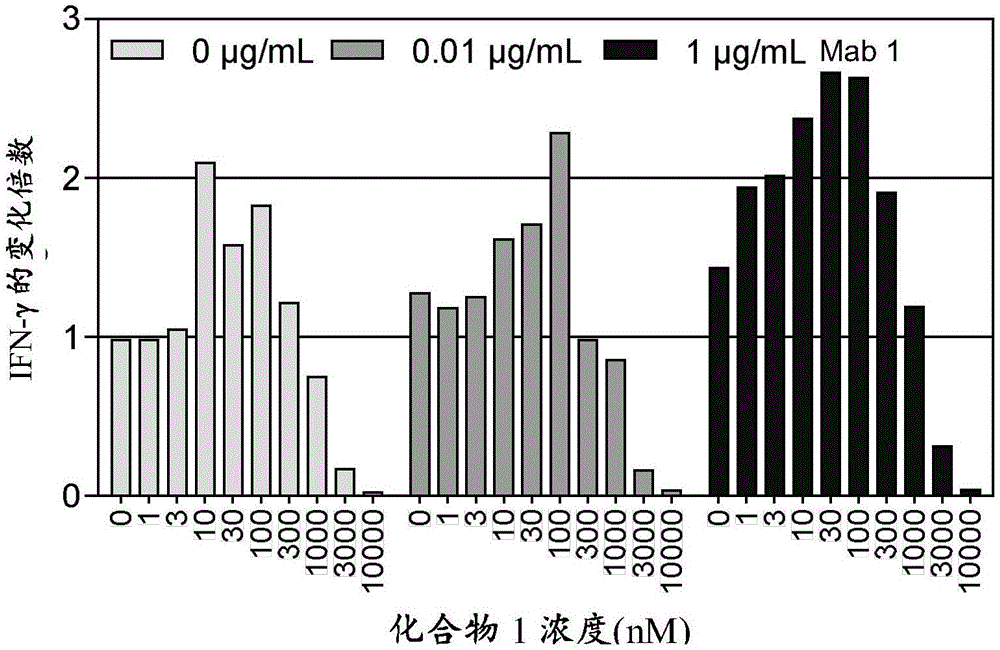
图1显示了用单独的raf抑制剂或用raf抑制剂与抗pd-1mab的组合进行治疗后肿瘤球体/pbmc共培养体系中活化的pbmc所产生的ifn-γ的水平。
图2显示了raf抑制剂和抗pd-1mab在人pbmc存在下对calu-6异种移植物模型中肿瘤生长的组合作用。
图3显示了raf抑制剂和抗pd-1mab在人pbmc存在下对人原发性结肠癌bcco-028异种移植物模型中肿瘤生长的组合作用。
图4显示了呈结晶形式(从异丙醇/水中结晶)的化合物1的x射线衍射图。
图5显示了化合物1的结晶形式的1h-nmr谱。
图6显示了化合物1的结晶形式的13c-nmr谱。
具体实施方式
缩写
在本申请公开的详细描述和实施例中将使用以下缩写:
定义
除非本申请另有具体定义,本申请使用的所有其它技术和科学术语具有本申请所属领域普通技术人员通常理解的含义。
除非上下文另有明确指示,本申请(包括所附权利要求书)使用的词语的单数形式诸如“一个”、“一种”和“该”包括其相应的复数指代。
在定量测量的上下文中使用的术语“约”是指标示量±20%或标示量±10%或±5%或±1%。例如,对于化合物1,约1的摩尔比(游离碱/马来酸,n)可在0.8和1.2之间变化。
本申请中的术语“烷基”是指单价支链或直链饱和烃链,其包含1-18个诸如1-12个进一步诸如1-6个碳原子。烷基包括但不限于甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正己基、正癸基、十四烷基等。
本申请中的术语“烯基”是指单价支链或直链不饱和烃基,其包含至少一个c=c双键和2-18个诸如2-8个进一步诸如2-6个碳原子。烯基包括但不限于乙烯基(即-ch=ch2)、1-丙烯基(或烯丙基即-ch2ch=ch2)、异丙烯基(-c(ch3)=ch2)等。
本申请中的术语“炔基”是指单价支链或直链不饱和烃基,其包含至少一个c≡c叁键和2-18个诸如2-8个进一步诸如2-6个碳原子。炔基包括但不限于乙炔基(-c≡ch)、炔丙基(或丙炔基即-c≡cch3)等。
本申请中的术语“环烷基”是指环状烷基,其包含3-20个碳原子或3-10个碳原子或3-8个碳原子或3-6个碳原子,其具有单环或多个稠环。环烷基可为饱和和部分不饱和的。单环饱和环烷基的实例包括但不限于选自环丙基、环丁基、环戊基、环己基、环庚基和环辛基的c3-8环烷基。单环部分不饱和环烷基的实例包括但不限于环戊-1-烯基、1-环戊-2-烯基、1-环戊-3-烯基、1-环己-1-烯基、1-环己-2-烯基、1-环己-3-烯基和环己二烯基。
本申请中的术语“芳基”是指通过由母体芳族环系的一个碳原子除去一个氢原子而衍生的包含6-20个碳原子诸如6-10个碳原子的单价芳族烃基。芳基包括二环基团,其包含与饱和、部分不饱和或芳族碳环或杂环稠合的芳环。芳基的实例包括但不限于衍生自苯(苯基)、经取代的苯、萘、蒽、联苯、茚、茚满、1,2-二氢萘、1,2,3,4-四氢萘等的基团。
本申请中的术语“卤素”或“卤代”是指f、cl、br或i。
本申请中的术语“杂芳基”是指选自以下的基团:
5-7元芳族单环,其包含至少一个例如1-4个或在一些实施方案中1-3个选自n、o和s的杂原子且其余环原子为碳;
8-12元二环,其包含至少一个例如1-4个或在一些实施方案中1-3个或在其它实施方案中1或2个选自n、o和s的杂原子且其余环原子为碳且其中至少一个环为芳族的且在芳族环中存在至少一个杂原子;及
11-14元三环,其包含至少一个例如1-4个或在一些实施方案中1-3个或在其它实施方案中1或2个选自n、o和s的杂原子且其余环原子为碳且其中至少一个环为芳族的且在芳族环中存在至少一个杂原子。
例如,杂芳基包括与5-7元环烷基环稠合的5-7元杂环芳环。就其中仅一个环包含至少一个杂原子的上述稠合二环杂芳基环系而言,连接点可位于杂芳环或环烷基环。
当杂芳基中s和o原子的总数超过1时,这些杂原子不彼此相邻。在一些实施方案中,杂芳基中s和o原子的总数不大于2。在一些实施方案中,芳族杂环中s和o原子的总数不大于1。
杂芳基的实例包括但不限于(由优先指定为1的连接位置开始编号)吡啶基(诸如2-吡啶基、3-吡啶基或4-吡啶基)、噌啉基、吡嗪基、2,4-嘧啶基、3,5-嘧啶基、2,4-咪唑基、咪唑并吡啶基、异噁唑基、噁唑基、噻唑基、异噻唑基、噻二唑基、四唑基、噻吩基、三嗪基、苯并噻吩基、呋喃基、苯并呋喃基、苯并咪唑基、吲哚基、异吲哚基、二氢吲哚基、酞嗪基、吡嗪基、哒嗪基、吡咯基、三唑基、喹啉基、异喹啉基、吡唑基、吡咯并吡啶基(诸如1h-吡咯并[2,3-b]吡啶-5-基)、吡唑并吡啶基(诸如1h-吡唑并[3,4-b]吡啶-5-基)、苯并噁唑基(诸如苯并[d]噁唑-6-基)、蝶啶基、嘌呤基、1-氧杂-2,3-二唑基、1-氧杂-2,4-二唑基、1-氧杂-2,5-二唑基、1-氧杂-3,4-二唑基、1-硫杂-2,3-二唑基、1-硫杂-2,4-二唑基、1-硫杂-2,5-二唑基、1-硫杂-3,4-二唑基、呋咱基、苯并呋咱基、苯并噻吩基、苯并噻唑基、苯并噁唑基、喹唑啉基、喹喔啉基、二氮杂萘基、呋喃并吡啶基、苯并噻唑基(诸如苯并[d]噻唑-6-基)、吲唑基(诸如1h-吲唑-5-基)和5,6,7,8-四氢异喹啉基。
本申请中的术语“杂环”或“杂环基”是指选自4-12元单环、二环和三环饱和和部分不饱和环的环,其除至少一个诸如1-4个进一步诸如1-3个进一步诸如1或2个选自氧、硫和氮的杂原子外还包含至少一个碳原子。本申请中的“杂环”还指与5、6和/或7元环烷基、碳环芳环或杂芳环稠合的包含至少一个选自n、o和s的杂原子的5-7元杂环,条件是当杂环与碳环芳环或杂芳环稠合时,连接点位于杂环,而当杂环与环烷基稠合时,连接点可位于环烷基或杂环。本申请中的“杂环”还指包含至少一个选自n、o和s的杂原子的脂族螺环,条件是连接点位于杂环。所述环可为饱和的或具有至少一个双键(即部分不饱和)。杂环可取代有氧代。连接点可为杂环中的碳或杂原子。杂环不是本申请定义的杂芳基。
杂环的实例包括但不限于(由优先指定为1的连接位置开始编号)1-吡咯烷基、2-吡咯烷基、2,4-咪唑烷基、2,3-吡唑烷基、1-哌啶基、2-哌啶基、3-哌啶基、4-哌啶基、2,5-哌嗪基、吡喃基、2-吗啉基、3-吗啉基、氧杂环丙基、氮杂环丙基、硫杂环丙基、氮杂环丁基、氧杂环丁基、硫杂环丁基、1,2-二硫杂环丁基、1,3-二硫杂环丁基、二氢吡啶基、四氢吡啶基、硫吗啉基、氧硫杂环己基、哌嗪基、高哌嗪基、高哌啶基、氮杂环庚基、氧杂环庚基、硫杂环庚基、1,4-氧硫杂环己基、1,4-二氧杂环庚基、1,4-氧硫杂环庚基、1,4-氧氮杂环庚基、1,4-二硫杂环庚基、1,4-硫氮杂环庚基、1,4-二氮杂环庚基、1,4-二噻烷基、1,4-氮硫杂环己基、氧氮杂
本申请中的术语“稠环”是指多环环系例如二环或三环环系,其中两个环仅共享两个环原子和一个键。稠环的实例可包括稠合二环环烷基环,诸如具有7-12个环原子的那些二环环烷基环,其排列成选自上述[4,4]、[4,5]、[5,5]、[5,6]和[6,6]环系的二环;稠合二环芳基环,诸如上述7-12元二环芳基环系;稠合三环芳基环,诸如上述10-15元三环芳基环系;稠合二环杂芳基环,诸如上述8-12元二环杂芳基环;稠合三环杂芳基环,诸如上述11-14元三环杂芳基环;及上述稠合二环或三环杂环基环。
本申请中的术语“施用”、“给药”、“治疗”和“处置”当应用于动物、人类、实验受试者、细胞、组织、器官或生物流体时是指外源性药物、治疗剂、诊断剂或组合物与动物、人类、受试者、细胞、组织、器官或生物流体的接触。对细胞的处置包括试剂与细胞的接触及试剂与流体的接触,其中所述流体与细胞接触。术语“施用”、“给药”、“治疗”和“处置”也指用试剂、诊断剂、结合化合物或另一个细胞进行的体外和离体处置,例如对细胞的体外和离体处置。本申请中的术语“受试者”包括任何生物体,优选动物,更优选哺乳动物(例如大鼠、小鼠、犬、猫、兔)且最优选人类。
本申请中的术语“药用盐”包括但不限于与无机酸的盐,其选自例如盐酸盐、磷酸盐、磷酸二氢盐、氢溴酸盐、硫酸盐、亚硫酸盐和硝酸盐;及与有机酸的盐,其选自例如苹果酸盐、马来酸盐、富马酸盐、酒石酸盐、琥珀酸盐、枸橼酸盐、乳酸盐、甲磺酸盐、对甲苯磺酸盐、2-羟基乙磺酸盐、苯甲酸盐、水杨酸盐、硬脂酸盐、烷酸盐诸如乙酸盐和与其中n选自0-4的hooc-(ch2)n-cooh的盐。类似地,药用阳离子的实例包括但不限于钠、钾、钙、铝、锂和铵。
本申请中的术语“抗体”在最广意义下使用且具体包括抗体(包括全长单克隆抗体)及抗体片段,只要其识别pd-1即可。抗体分子通常为单特异性,但也可描述为独特特异性、异种特异性或多特异性。抗体分子通过特异性结合位点与抗原上的特异性抗原决定簇或表位结合。“抗体片段”包含全长抗体的一部分,通常为全长抗体的抗原结合或可变区。抗体片段的实例包括fab、fab’、f(ab’)2和fv片段;双抗体;线性抗体;单链抗体分子;和由抗体片段形成的多特异性抗体。
本申请中的术语“单克隆抗体”或“mab”或“mab”是指基本上均质的抗体群,即包含在群中的抗体分子除可少量存在的可能天然突变外在氨基酸序列方面是相同的。相反地,常规(多克隆)抗体制品通常包含多种不同的抗体,所述多种不同的抗体在其可变结构域特别是其cdr中具有不同的氨基酸序列,所述常规(多克隆)抗体制品通常对不同的表位都具有特异性。修饰语“单克隆”表示由基本上均质的抗体群获得的抗体的特征且不应被解释为需要通过任何特定方法产生抗体。单克隆抗体(mab)可通过本领域技术人员已知的方法获得。参见例如kohler等人(1975);u.s.专利4,376,110;ausubel等人(1987-1999);harlow等人(1988);和colligan等人(1993)。本申请公开的mab可为任何免疫球蛋白类别,包括igg、igm、igd、ige、iga及其任何亚类。可对产生mab的杂交瘤进行体外或体内培养。体内产生可获得高滴度的mab,其中将来自各个杂交瘤的细胞腹膜内注射到小鼠(诸如原始致敏balb/c小鼠)中以产生含有高浓度所需mab的腹水液。可使用本领域技术人员已知的柱色谱法由上述腹水液或培养物上清液纯化同种型igm或igg的mab。
通常,基本抗体结构单元包含四聚体。每个四聚体包含两对相同的多肽链,每对具有一条“轻链”(约25kda)和一条“重链”(约50-70kda)。每条链的氨基末端部分包含具有约100-110个或更多个氨基酸的主要负责抗原识别的可变区。重链的羧基末端部分可限定主要负责效应子功能的恒定区。通常,人轻链被分类为κ和λ轻链。另外,人重链通常被分类为α、δ、ε、γ或μ且将抗体的同种型分别限定为iga、igd、ige、igg和igm。在轻链和重链中,可变区和恒定区通过具有约12个或更多个氨基酸的“j”区连接,其中重链还包含具有约10个或更多个氨基酸的“d”区。
每个轻链/重链(vk/vh)对的可变区形成抗体结合位点。因此,完整抗体通常具有两个结合位点。除双功能或双特异性抗体外,两个结合位点通常是相同的。
通常,重链和轻链的可变结构域都包含位于相对保守的框架区(fr)中的三个高变区(也称为“互补决定区(cdr)”)。cdr通常通过框架区来对齐,由此能够结合特异性表位。通常,从n末端到c末端,轻链和重链可变结构域都包含fr-1、cdr-1、fr-2、cdr-2、fr-3、cdr-3和fr-4。通常,根据以下文献的定义将氨基酸指派至各个结构域:sequencesofproteinsofimmunologicalinterest,kabat等人,nationalinstitutesofhealth,bethesda,md.,5<m>ed.,nihpubl.no.91-3242(1991);kabat(1978)adv.prot.chem.32:1-75;kabat等人(1977)j.biol.chem.252:6609-6616;chothia等人(1987)jmol.biol.196:901-917;或chothia等人(1989)nature342:878-883。
术语“高变区”是指抗体的负责抗原结合的氨基酸残基。高变区包含来自“互补决定区”或“cdr”(即轻链可变域中的cdr-l1、cdr-l2和cdr-l3及重链可变域中的cdr-h1、cdr-h2和cdr-h3)的氨基酸残基。参见kabat等人(1991)sequencesofproteinsofimmunologicalinterest,5thed.publichealthservice,nationalinstitutesofhealth,bethesda,md.(通过序列来定义抗体的cdr区);另见chothiaandlesk(1987)j.mol.biol.196:901-917(通过结构来定义抗体的cdr区)。术语“框架”或“fr”残基是指除本申请定义为cdr残基的高变区残基外的那些可变结构域残基。
除非另有说明,“抗体片段”或“抗原结合片段”是指抗体的抗原结合片段即保留与由全长抗体结合的抗原特异性结合的能力的抗体片段例如保留一个或多个cdr区的片段。抗体结合片段的实例包括但不限于fab、fab’、f(ab’)2和fv片段;双抗体;线性抗体;单链抗体分子,例如sc-fv;纳米抗体;和由抗体片段形成的多特异性抗体。
与特定靶标蛋白“特异性结合”的抗体是与其它蛋白质相比优先与该靶标结合的抗体,但是该特异性不需要绝对结合特异性。若抗体的结合在例如不产生不期望的结果诸如假阳性的情况下决定了样品中靶标蛋白的存在,则抗体被认为就其所预期的靶标而言是“特异性的”。可用于本申请的抗体或其结合片段将以比与非靶标蛋白的亲和力大至少2倍、优选大至少10倍、更优选大至少20倍且最优选大至少100倍的亲和力与靶标蛋白结合。本申请抗体被描述为与包含给定氨基酸序列的多肽例如成熟人pd-1分子的氨基酸序列特异性结合,条件是其与包含该序列的多肽结合,但是不与不具有该序列的蛋白质结合。
本申请中的术语“人抗体”是指仅包含人免疫球蛋白序列的抗体。若在小鼠、小鼠细胞或衍生自小鼠细胞的杂交瘤中产生,则人抗体可含有鼠型糖链。类似地,“小鼠抗体”或“大鼠抗体”是指分别仅包含小鼠或大鼠免疫球蛋白序列的抗体。
术语“人源化抗体”是指以下抗体形式,其含有来自非人类(例如鼠类)抗体及人抗体的序列。上述抗体含有衍生自非人类免疫球蛋白的小序列。通常,人源化抗体将包含基本上所有至少一个且通常为两个可变结构域,其中所有或基本上所有高变环与非人类免疫球蛋白的那些高变环是对应的且所有或基本上所有fr区是人免疫球蛋白序列的那些fr区。人源化抗体任选还将包含免疫球蛋白恒定区(fc)通常为人免疫球蛋白恒定区(fc)的至少一部分。当必要时,将前缀“hum”、“hu”或“h”加至抗体克隆名称以区分人源化抗体与亲本啮齿动物抗体。啮齿动物抗体的人源化形式通常将包含与亲本啮齿动物抗体相同的cdr序列,尽管可包括某些氨基酸取代以增加亲和力、增加人源化抗体的稳定性或出于其它原因。
本申请中的术语“癌症”或“肿瘤”是指或描述哺乳动物中通常以不受调节的细胞生长为特征的生理状态。癌症的实例包括但不限于肾上腺癌、膀胱癌、骨癌、脑癌、乳腺癌、子宫颈癌、结肠直肠癌、子宫内膜癌、头颈癌、肾癌、肝癌、肺癌(包括小细胞肺癌或非小细胞肺癌)、淋巴瘤、黑素瘤、卵巢癌、胰腺癌、皮肤癌或甲状腺肿瘤及其并发症。可通过本申请公开的组合来治疗的特别优选的癌症或肿瘤包括以b-raf突变、k-ras/n-ras突变和/或nf1突变为特征的那些癌症或肿瘤。可通过本申请公开的组合来治疗的最优选的癌症或肿瘤包括各自与k-ras突变相关的非小细胞肺癌、结肠直肠癌和子宫内膜癌。
除非另有说明,术语“cdr”是指免疫球蛋白可变区中使用kabat编号系统来定义的互补决定区。
“pd-1拮抗剂”是指阻断在癌细胞上表达的pd-l1与在免疫细胞(t细胞、b细胞或nkt细胞)上表达的pd-1的结合且优选还阻断在癌细胞上表达的pd-l2与免疫细胞所表达的pd-1的结合的任何化合物或生物分子。pd-1及其配体的替代名称或同义词包括:对于pd-1,包括pdcd1、pd1、cd279和sleb2;对于pd-l1,包括pdcd1l1、pdl-1、b7h1、b7-4、cd274和b7-h;且对于pd-l2,包括pdcd1l2、pdl2、b7-dc、btdc和cd273。
pd-1拮抗剂
如上述五个方面各自所公开,pd-1拮抗剂为与人pd-1特异性结合的抗体或其抗原结合片段。
如上述五个方面各自所公开,pd-1拮抗剂为包含重链可变区(vh)和轻链可变区(vk)的抗体,所述重链可变区(vh)和轻链可变区(vk)含有下列互补决定区(cdr):
如上述五个方面各自所公开,pd-1拮抗剂为包含重链可变区(vh)和轻链可变区(vk)的抗体,所述重链可变区(vh)和轻链可变区(vk)含有下列cdr的任意组合:
如上述五个方面各自所公开,pd-1拮抗剂为包含重链可变区(vh)和轻链可变区(vk)的抗体,所述重链可变区(vh)和轻链可变区(vk)包含:
如上述五个方面各自所公开,pd-1拮抗剂为含有igg4重链效应子或恒定域的抗体,所述igg4重链效应子或恒定域包含seqidno:83-88中的任何一种。
如上述五个方面各自所公开,pd-1拮抗剂为含有f(ab)或f(ab)2的抗体,所述f(ab)或f(ab)2包含上述结构域,包括重链可变区(vh)、轻链可变区(vk)和igg4重链效应子或恒定域。
如上述五个方面各自所公开,pd-1拮抗剂为包含重链可变区(vh)和轻链可变区(vk)及igg4重链效应子或恒定域的抗体,所述igg4重链效应子或恒定域包含seqidno:87或88,其中所述重链可变区(vh)和轻链可变区(vk)包含:
如上述五个方面各自所公开,pd-1拮抗剂为包含重链可变区(vh)和轻链可变区(vk)及igg4重链效应子或恒定域的抗体,所述igg4重链效应子或恒定域包含seqidno:88,其中所述重链可变区(vh)和轻链可变区(vk)分别包含seqidno:24和seqidno:26。
本申请公开的抗pd1抗体及其抗体片段可根据wo2015/035606a1所公开的内容来制备,通过引用将其全部内容明确并入本申请。
raf抑制剂
“raf抑制剂”是指式(i)化合物或其立体异构体或其药用盐。
如上述五个方面各自所公开,raf抑制剂为式(i)化合物或其立体异构体或其药用盐:
其中
x选自ch2和o;
r8、r9、r10和r11可相同或不同,各自选自氢、卤素、烷基、烯基、环烷基、芳基、杂环基、杂芳基、炔基、-nr13r14、-or13、-cor13、-co2r13、-conr13r14、-c(=nr13)nr14r15、-nr13cor14、-nr13conr14r15、-nr13co2r14、-so2r13、-so2芳基、-nr13so2nr14r15和-nr13so2r14,其中所述烷基、烯基、炔基、环烷基、杂芳基、芳基和杂环基各自任选取代有至少一个取代基r16;或(r8和r9)和/或(r9和r10)和/或(r10和r11)与和它们连接的环一起形成选自杂环基和杂芳基环的稠环,其任选取代有至少一个取代基r16;
r13、r14和r15可相同或不同,各自选自h、卤代烷基、烷基、烯基、炔基、环烷基、杂环基、芳基和杂芳基;或(r13和r14)和/或(r14和r15)与和它们连接的原子一起各自形成选自杂环基和杂芳基环的环,其取代有至少一个取代基r16;
r16选自卤素、卤代烷基、烷基、烯基、环烷基、芳基、杂芳基、杂环基、炔基、氧代、-cn、-or’、-nr’r”、-cor’、-co2r’、-conr’r”、-c(=nr’)nr”r”’、-nr’cor”、-nr’conr’r”、-nr’co2r”、-so2r’、-so2芳基、-nr’so2nr”r”’、-nr’o2r”和-nr’so2芳基,其中r’、r”和r”’独立选自h、卤代烷基、烷基、烯基、炔基、环烷基、杂环基、芳基和杂芳基;或(r’和r”)和/或(r”和r”’)与和它们连接的原子一起形成选自杂环基和杂芳基环的环。
在一些实施方案中,式(i)化合物为光学纯的。
在一些实施方案中,式(i)中的x为o。
在一些实施方案中,式(i)中的x为ch2。
在一些实施方案中,式(i)中的r8、r9、r10和r11可相同或不同,各自独立选自烷基(例如甲基、叔丁基)、氢、卤代烷基(例如-cf3)、卤素、羟基、-cn、-o烷基(例如烷氧基)、-o卤代烷基(例如ocf3)和芳基(例如苯基)。
如上述五个方面各自所公开,raf抑制剂为式(i)化合物或其立体异构体或其药用盐:
其中
x选自ch2和o;
r8、r9、r10和r11可相同或不同,各自选自氢、卤素、烷基、-cn、环烷基、芳基、杂环基、-or13、-conr13r14,其中所述烷基和芳基各自任选取代有至少一个取代基r16;或(r8和r9)和/或(r9和r10)和/或(r10和r11)与和它们连接的环一起形成选自环烷基的稠环;
r13和r14可相同或不同,各自选自h、烷基和卤代烷基;
r16选自卤素、卤代烷基和烷基。
如上述五个方面各自所公开,raf抑制剂为式(i)化合物或其立体异构体或其药用盐:
其中
x选自ch2和o;
r8选自h、f、cl和br;
r9选自h、f、cl、br和c1-6烷基;
r10选自h、f、cl、br、oh、-cn、c1-6烷基、cf3、苯基、oc1-6烷基、oc1-6卤代烷基和-conr13r14,其中r13和r14可相同或不同,各自选自h和c1-6烷基;
r11选自h、f、cl、br和cf3;
或(r9和r10)与和它们连接的环一起形成选自c5-6环烷基的稠环。
如上述五个方面各自所公开,raf抑制剂为选自以下化合物的化合物或其立体异构体或其药用盐:
如上述五个方面各自所公开,raf抑制剂为选自以下化合物的化合物或其立体异构体或其药用盐:
如上述五个方面各自所公开,raf抑制剂为选自以下化合物的化合物或其药用盐:
如上述五个方面各自所公开,raf抑制剂为式(ii)化合物或其药用盐:
如上述五个方面各自所公开,raf抑制剂为式(ii)化合物的马来酸盐。
如上述五个方面各自所公开,raf抑制剂为式(iii)化合物:
其中n为约0.5至约1.5的数字。
如上述五个方面各自所公开,raf抑制剂为式(iiia)化合物即化合物1:
本申请公开的raf抑制剂诸如式(i)化合物可通过wo2013/097224a1所公开的合成路线来合成,通过引用将其全部内容明确并入本申请。本申请公开的raf抑制剂即化合物1可根据pct/cn2016/079251中的操作来制备,通过引用将其全部内容明确并入本申请。
组合疗法
组合疗法可按同时或分开或依序方案来施用。当依序施用时,组合可按两次或更多次给药来施用。组合施用包括使用分开的制剂共同施用和以任意顺序连续施用,其中优选存在两种(或所有)活性剂同时发挥其生物活性的时段。
在上述五个方面各自的一个实施方案中,raf抑制剂或其药用盐可在施用pd-1拮抗剂后施用约1至约10天的时段。在上述五个方面各自的另一个实施方案中,raf抑制剂或其药用盐可在组合施用开始前施用约1至10天的时段。在上述五个方面各自的另一个实施方案中,raf抑制剂或其药用盐的施用和pd-1拮抗剂的施用在同一天开始。
任何上述共同施用的药剂的合适剂量是目前使用的那些剂量且可由于raf抑制剂和pd-1拮抗剂的组合作用(协同作用)诸如增加的治疗指数或减轻的毒性或其它副作用或后果而降低。
在抗癌疗法的具体实施方案中,raf抑制剂和pd-1拮抗剂可进一步与手术疗法和放射疗法组合。
在上述五个方面各自的实施方案中,本申请公开的raf抑制剂和pd-1拮抗剂的量及施用的相对时间安排通过待治疗的患者的个体需要、施用途径、疾病或病痛的严重程度、给药时间表及指定医生所作出的评价和判断来确定。
例如,raf抑制剂的施用剂量为1-100mg/天(以母体化合物计)且施用频率为每天一至三次;优选地,raf抑制剂的施用剂量为5-80mg/天(以母体化合物计)且施用频率为每天一至三次;更优选地,raf抑制剂的施用剂量为10-40mg/天(以母体化合物计)且施用频率为一天一次。然而,基于活性化合物,本申请公开的raf抑制剂的有效剂量的优选范围可为约0.01-10mg/天/千克体重或更优选0.1-1mg/天/千克体重,在单一或分开剂量中。在一些情况下,应用上述剂量范围的下限是较合适的,而在其它情况下,可使用较高的剂量而不引起有害的副作用。在上述五个方面各自的一些优选实施方案中,raf抑制剂以5-80mg的剂量每天施用一次。
pd-1拮抗剂以0.5-30mg/kg诸如0.5-20mg/kg进一步诸如0.5-10mg/kg的剂量每周一次或每两周一次或每三周一次或每四周一次施用。
本申请公开的raf抑制剂和pd-1拮抗剂可按多种已知方式来施用,诸如口服、局部、经直肠、胃肠外、通过吸入喷雾或经由所植入的储库,尽管任何给定情况下最合适的途径将取决于具体的宿主及施用活性成分所针对的病症的性质和严重程度。本申请使用的术语“胃肠外”包括皮下、皮内、静脉内、肌内、关节内、动脉内、滑膜内、胸骨内、鞘内、病灶内和颅内注射或输注技术。
在上述五个方面各自的一个实施方案中,本申请公开的raf抑制剂和pd-1拮抗剂可按不同途径来施用。在优选实施方案中,raf抑制剂口服施用,而pd-1拮抗剂胃肠外诸如皮下、皮内、静脉内或腹膜内施用。
在上述五个方面各自的实施方案中,pd-1拮抗剂为包含重链可变区(vh)和轻链可变区(vk)及igg4重链效应子或恒定域的抗体,所述igg4重链效应子或恒定域包含seqidno:88,其中所述重链可变区(vh)和轻链可变区(vk)分别包含seqidno:24和seqidno:26;且raf抑制剂为本申请公开的式(iiia)化合物。
在上述五个方面各自的实施方案中,pd-1拮抗剂mab-1以0.5-10mg/kg的剂量每周一次(qw)或每两周一次(q2w)或每三周一次(q3w)向受试者静脉内(i.v.)或腹膜内(i.p.)施用,而raf抑制剂化合物1以5-80mg的剂量每天一次(qd)向受试者施用。在一些优选实施方案中,pd-1拮抗剂mab-1以0.5-10mg/kg的剂量每周一次(qw)或每两周一次(q2w)或每三周一次(q3w)向受试者静脉内(i.v.)或腹膜内(i.p.)施用,而raf抑制剂化合物1以10-30mg的剂量每天一次(qd)向受试者施用。在更优选的实施方案中,pd-1拮抗剂mab-1胃肠外诸如皮下、皮内、静脉内或腹膜内施用。
治疗方法
所述药物组合在抑制癌症的肿瘤生长中产生协同功效,所述癌症为诸如肾上腺癌、膀胱癌、骨癌、脑癌、乳腺癌、子宫颈癌、结肠直肠癌、子宫内膜癌、头颈癌、肾癌、肝癌、肺癌(包括小细胞肺癌或非小细胞肺癌)、淋巴瘤、黑素瘤、卵巢癌、胰腺癌、皮肤癌或甲状腺肿瘤及其并发症。具体地,所述组合在与b-raf突变、k-ras/n-ras突变和/或nf1突变相关的上述癌症中是有效的。可通过本申请公开的组合来治疗的最优选的癌症或肿瘤包括各自与k-ras突变相关的非小细胞肺癌、结肠直肠癌和子宫内膜癌。所述组合可在用于预防癌症、延缓癌症进展或治疗癌症的方法中使用。
实施例
实施例a:化合物1(即倍半马来酸盐)的制备
步骤1:中间体1的合成
在氮气保护下在内温≤35℃向etona(154kg)于dmf(989kg)中的搅拌的溶液中添加etsh(68.6kg)。将混合物在内温≤35℃搅拌60-90min。添加5-甲氧基苯并呋喃(58.75kg)于dmf(55.0kg)中的溶液。将混合物加热至110-130℃,搅拌45小时,然后在低于90℃真空浓缩。将混合物冷却至10-20℃后,逐滴添加2nhcl(1326kg),然后在内温≤35℃添加etoac(531kg)和h2o2(129kg)。将混合物搅拌30-60min。分离有机层后,水相用etoac萃取。合并的有机相用饱和盐水洗涤两次,然后将溶剂蒸发至干。在低于40℃将meoh和naoh(44.5kg)于水(185kg)中的溶液逐滴加到残余物中。将混合物在30-40℃搅拌5-7小时。添加用水(77kg)湿润的活性碳(74kg)。将混合物在30-40℃搅拌4-6小时并过滤;且滤饼用meoh和水洗涤。将dcm加到滤液中并在低于40℃用35%hcl水溶液将ph调节至1。水相用dcm萃取且有机相用25%nacl洗涤且在低于40℃浓缩。残余物直接用于下一步。1hnmr(400mhz,dmso-d6)δ9.14(s,1h),7.86(d,j=2.0hz,1h),7.36(d,j=8.8hz,1h),6.94(d,j=2.4hz,1h),6.79(dd,j=2.0,0.9hz,1h),6.74(dd,j=8.8,2.4hz,1h)ppm。ms:m/e135(m+1)+。
步骤2:中间体2的合成
在-5至0℃向苯并呋喃-5-醇(中间体1,33.1kg)和et3n(50.8kg)于dcm(155kg)中的搅拌的溶液中逐滴添加tmscl(30.4kg)于dcm(50kg)中的溶液。将混合物温热到0至10℃并在该温度搅拌2小时(ipc检查int-1/int-2=37.4%)。将混合物冷却到-5至0℃并逐滴添加tmscl(10.6kg)于dcm(8kg)中的溶液,然后将混合物温热到0至10℃并在该温度搅拌1h。将混合物在低于40℃浓缩并向混合物中添加正庚烷。将混合物搅拌20-30min且过滤且滤饼用正庚烷洗涤。从滤液中蒸馏出溶剂,得到粗中间体2(int-2%:62.7%,kf:0.01%)。在-5至0℃向上述粗中间体2和et3n(8.6kg)于dcm(149kg)中的搅拌的溶液中逐滴添加tmscl(9.0kg)于dcm(10kg)中的溶液。将混合物温热到0至10℃并在该温度搅拌1h(tlc显示反应完成)。将反应混合物在低于40℃浓缩并向混合物中添加正庚烷。将混合物搅拌20-30min,然后过滤且滤饼用正庚烷洗涤。从滤液中蒸馏出溶剂,得到中间体2(41.5kg,int-2%:98.1%),其为无色油状物。1hnmr(400mhz,dmso-d6)δ7.69(d,j=2.0hz,1h),7.21(d,j=8.8hz,1h),6.84(d,j=2.5hz,1h),6.61(d,j=2.0hz,1h),6.56(dd,j=8.8,2.5hz,1h),0.00(s,9h)ppm。
步骤3:中间体3的合成
在环境温度在n2气氛下将三氟甲磺酸亚铜(i)(与甲苯的2:1络合物,0.41kg)和(s,s)-evans配体(0.552kg)在dcm(160kg)中搅拌1-2小时。添加中间体2(37.0kg),然后在20-30℃缓慢添加重氮基乙酸乙酯(58kg)于dcm(450kg)中的溶液。将反应混合物在20-30℃搅拌0.5-1h(ipc:int-2/int-3≤0.2%,残留的n2chco2et:0.05%≤1.0%)。在20-30℃历时40-50min将edta二钠溶液(0.05mol/l,150kg)分三次加到反应混合物中。有机相在20-30℃用25%nacl水溶液洗涤两次并在低于30℃浓缩。将残余物减压蒸馏并在120-144℃收集粗中间体3(36.26kg,84.5%)。该粗化合物包含内型对映异构体,其可在下一步中被除去。1hnmr(400mhz,dmso-d6)δ6.79(d,j=2.4hz,1h),6.59(d,j=8.4hz,1h),6.42(dd,j=8.4,2.4hz,1h),4.95(dd,j=5.4,1.0hz,1h),3.08(dd,j=5.4,3.2hz,1h),1.02(dd,j=3.1,1.2hz,1h),0.00(s,9h)ppm。
步骤4和5:中间体5和中间体6的合成
在20-30℃向中间体4(36.3kg)于meoh(108kg)中的溶液中添加hcl/meoh溶液(5m,0.11kg)且将混合物搅拌2-3小时(ipc:l/m:0.5%,手性纯度90.0%)。在20-30℃逐滴添加et3n(0.22kg)。将混合物浓缩且残余物用正庚烷/etoac(4:1)稀释,然后浓缩。将温度调节至10-20℃并在10-20℃搅拌2-4小时后,将混合物过滤,得到湿产物(中间体5:94.0%,手性纯度:90.5%)。1hnmr(400mhz,dmso-d6)δ9.05(s,1h),6.89(d,j=2.4hz,1h),6.72(d,j=8.4hz,1h),6.55(dd,j=8.4,2.4hz,1h),5.11(dd,j=5.4,1.0hz,1h),3.27(dd,j=5.4,3.0hz,1h),1.19-1.17(m,1h)ppm。粗产物用正庚烷/etoac(20:1)浆化三次,得到浅黄色固体,将其在40-50℃干燥12-16小时,得到16.55kg产物(中间体6:98.6%;手性纯度:99.3%)。
步骤6和7:中间体7和中间体8的合成
在40-60℃向中间体6(14kg)和5-氟-3,4-二氢-1,8-二氮杂萘-2(1h)-酮(sm2,11.2kg)于dmf(66kg)中的溶液中添加cs2co3(26kg)且将混合物温热至110-120℃且在110-120℃搅拌3小时。在25-35℃用乙酸(12.0kg)将反应混合物的ph调节至6。添加水(520kg)且将混合物搅拌1-2小时。过滤后,固体用ea(78kg)浆化,得到湿产物(纯度:(中间体7+中间体8)%:98%)。将氢氧化钠水溶液(125kg,2m)加到湿产物于thf(240kg)中的搅拌的溶液中并在20-30℃搅拌2-3小时(ipc:int-7/int-8:0.9%)。在20-30℃用4nhcl(37kg)将混合物的ph调节至4-5,然后搅拌0.5-1h。将混合物在低于50℃浓缩且固体从溶液中析出。过滤后,在35-45℃将湿产物在thf中再次浆化1-2小时,然后过滤。将所得湿产物在45-65℃干燥40小时,得到标题化合物中间体8(18.95kg:化学纯度99%,手性纯度100%)。1hnmr(400mhz,dmso-d6)δ12.59(s,1h),10.43(s,1h),7.92(d,j=5.8hz,1h),7.29(d,j=2.4hz,1h),6.97(d,j=8.8hz,1h),6.93(dd,j=8.8,2.4hz,1h),6.21(d,j=5.8hz,1h),5.21(dd,j=5.4,1.0hz,1h),3.27-3.25(m,1h),2.89(t,j=7.8hz,2h),2.51(d,j=8.8hz,2h),1.19(dd,j=3.0,1.0hz,1h)ppm。ms:m/e339(m+1)+。
步骤8:中间体9的合成
在0-15℃将中间体8(13.3kg)、dipea(16kg)和hatu(18.1kg)于dmf(167kg)中的溶液逐滴加到4-(三氟甲基)苯-1,2-二胺(sm3,7.6kg)于dmf(74kg)中的混合物中。将混合物在20-25℃搅拌4-6小时(ipc:int-9/int-9:未检测)。将活性碳(5.3kg)/dmf(7.5kg)加到反应混合物中,在40-45℃搅拌2-4小时,然后过滤。在15-30℃将水(846kg)逐滴加到滤液中且当搅拌1-2小时时,固体从溶液中析出。将析出物过滤并在20-30℃在etoh中浆化2-4小时。过滤后,将湿产物在45-60℃干燥37小时,得到标题化合物中间体9(17.60kg:95.5%)。
步骤9:化合物1的游离碱的合成
将中间体9(17kg)和水(1.5kg)于acoh(360kg)中的溶液在65-70℃搅拌20小时(ipc:r/s≤1.0%)。将混合物在低于55℃浓缩至干并向残余物中添加活性碳(17kg)及meoh(32kg)。将混合物在约50℃搅拌1h。过滤后,将滤液在低于45℃浓缩以除去溶剂。向残余物中添加ea(160kg)和水(330kg),然后在20-30℃添加naoh水溶液(2mol/l)直至ph为8-9。将有机层分离且水相用ea萃取。合并的有机相用水洗涤两次,浓缩至干,得到呈游离碱形式的化合物1。1h-nmr(600mhz,dmso-d6)δ12.84(s,1h),10.47(s,1h),7.98(d,j=5.8hz,1h),7.86(d,j=1.2hz,1h),7.69(m,1h),7.48(t,j=6.2hz,1h),7.38(d,j=2.6hz,1h),7.08(d,j=8.7hz,1h),7.02(dd,j=8.7,2.6hz,1h),6.29(d,j=5.8hz,1h),5.43(dd,j=5.4,1.2hz,1h),3.55(dd,j=5.3,3.3hz,1h),2.95(t,j=7.7hz,2h),2.55(t,j=7.7hz,2h),1.97(d,j=1.3hz,1h)ppm。
步骤10:化合物1的合成
向步骤9的残余物中添加ipa(83kg)。向混合物中添加马来酸(5kg)于水(29kg)中的溶液并在约50℃搅拌4小时,然后冷却至35℃并在该温度搅拌12小时。将所得固体过滤,在40-60℃干燥并在微粉磨中微粉化,得到白色固体(化合物1倍半马来酸盐,8.36kg),其具有d90=4.1μm、d10=1.5μm、d50=2.4μm的粒径。如图4所示,通过粉末x射线衍射图方法将化合物1鉴定为结晶形式。呈结晶形式的化合物1的1h-nmr谱如图5所示。呈结晶形式的化合物1的13c-nmr谱如图6所示。
实施例1
raf抑制剂与抗pd-1-mab的组合对体外3d球体/pbmc共培养模型中t细胞功能的作用
方法
为了生成肿瘤球体,将hek-293-os8-pd-l1细胞和sk-mel-28细胞(b-rafv600e)以1:1的比例以5×104个细胞/ml的密度在冰冷的培养基中混合。将
结果
单独的化合物1在共培养体系中在10nm至100nm的浓度促进ifn-γ从pbmc中释放,而在高浓度(≥3μm)抑制ifn-γ释放。中等水平的化合物1与mab1的组合显示出显著增加的pbmcifn-γ产生,这表明较好的体外t细胞活性(图1)。
实施例2
抗pd-1mab与raf抑制剂的组合在k-ras突变肺癌和b-rafv600e突变结肠癌模型中在人pbmc存在下的作用
方法
在植入当天,从健康志愿者所捐献的120ml血液中分离人外周血单核细胞(pbmc)。简言之,将外周血收集到含有肝素钠的真空采血管中。pbmc通过密度梯度离心使用histopaque-1077来分离且用dulbecco磷酸盐缓冲盐水(dpbs)洗涤一次。细胞团块用适当体积的dpbs混悬以使最终浓度为1×108个细胞/ml并在接种前置于冰上。nod/scid小鼠每天一次且持续两天用环磷酰胺(cyclophosphamide)(在盐水中制备,150mg/kg,i.p.)和双硫仑(disulfiram)(在0.8%吐温-80/盐水中制备,125mg/kg,p.o.,每剂环磷酰胺后1小时)预处理。然后在第二剂环磷酰胺后24小时,向动物注射肿瘤细胞或肿瘤片段和pbmc的混合物。
对于calu-6k-ras突变肺癌模型,将细胞在补充有10%(v/v)胎牛血清及100μg/ml青霉素和链霉素的dulbecco基本必需培养基(dmem)中培养。在植入当天,培养基用新鲜培养基替换。5小时后,除去培养基并如上所述收集细胞,除了将细胞重新混悬于冷的(4℃)dpbs中以使最终浓度为5×107个细胞/ml并在接种前置于冰上。以1:1:2的比例混合calu-6细胞、pbmc和基质胶(bd,目录号356237)。每只小鼠的右腋区用70%乙醇清洁,然后接种细胞。通过26号针头在右前胁向每只动物皮下注射5×106个calu-6细胞和2.5×106个pbmc(200μl细胞混合物在50%基质胶中)。
对于b-rafv600e突变结肠癌衍生自患者的异种移植物(pdx)模型,bcco-028衍生自由结肠癌患者手术取出的肿瘤组织。在患者手术后2-4小时内,将肿瘤样品皮下植入到经麻醉的nod/scid小鼠的肩胛区中。肿瘤生长至约300-1000mm3后,将肿瘤手术取出并通过随后的移植将片段在nod/scid小鼠中传代。在第二剂环磷酰胺后24小时,向动物植入bcco-028肿瘤片段和pbmc。每只nod/scid小鼠的右腋区用70%乙醇清洁,然后接种肿瘤片段。通过14号套针针头在右胁向每只动物皮下植入bcco-028结肠癌(第5代)的片段(约3×3×3mm3),然后通过26号针头在肿瘤片段的边缘附近皮下注射于200μl50%基质胶中的5×106个pbmc。
自接种细胞或片段后第0天开始,将动物随机分成4组,其中每组8-11只小鼠。这些组包括对照组(无药物治疗)、5mg/kg化合物1组(基于游离碱重量)、10mg/kgmab1组和化合物1+mab1组合组。治疗以10ml/kg体重的体积施用,在给药前即刻评估并相应地调整给药体积。化合物1通过经口管饲法(p.o.)每天一次(qd)施用,而mab1通过腹膜内(i.p.)注射每周一次(qw)施用。植入后,初始肿瘤体积使用卡尺在两个维度测量。
每周两次记录个体体重及在研究期间每天监测小鼠的毒性临床体征。一旦其肿瘤体积达到2,500mm3、肿瘤发生溃疡或体重减轻超过20%,就使用二氧化碳对小鼠进行安乐死。
肿瘤体积使用以下公式来计算:v=0.5×(a×b2),其中a和b分别为肿瘤的长径和短径。肿瘤生长抑制(tgi)使用以下公式来计算:
%tgi=100×[1-(治疗t/安慰剂t)]
治疗t=时间t时的经治疗的肿瘤体积
安慰剂t=时间t时的安慰剂肿瘤体积
结果
在nod/scid小鼠中在人pbmc存在下皮下生长的bcco-028pdx和calu-6细胞中检查化合物1和mab1的体内功效。化合物1针对裸鼠中的pdxbcco-028结肠直肠癌(b-rafv600e突变)和人calu-6肺腺癌(k-ras突变)肿瘤异种移植物具有显著的抗肿瘤活性。另外,如下图所示,化合物1和mab1的协同功效通过这些模型中的肿瘤生长曲线而得以清楚的显示。组合治疗组中的肿瘤显著小于任何单项治疗(图2和图3)。
上述实施例和对某些实施方案的描述应被认为是说明性的而非限制权利要求书所定义的本申请。容易理解的是,可在不脱离权利要求书所描述的本申请的情况下使用上述特征的多种变化和组合。所有这些变化都意欲包括在本申请的范围内。所引用的所有参考文献通过引用整体并入本申请。
应理解的是,在本申请引用任何现有技术出版物的情况下,该引用不意味着承认该出版物在任何国家或地区构成本领域常规知识的一部分。
在本申请下述权利要求书和上述说明书中,除非上下文出于语言清楚或含义必要的目的而另有要求,词语“包括”、“包含”或“含有”以包括性含义使用即表明存在所描述的特征,但是不排除在本申请各个实施方案中存在或添加其它特征。
本申请通过指明出处而引用的所有出版物、专利、专利申请和公开的专利申请所公开的内容通过引用整体并入本申请。
- 还没有人留言评论。精彩留言会获得点赞!