蛋白质检索模型构建方法、检索方法、设备和存储介质与流程
本申请涉及生物信息学和计算机生物学技术领域,具体涉及一种蛋白质检索模型构建方法、检索方法、设备和存储介质。
背景技术:
蛋白质相似性分析已经成为生物信息学和计算机生物学的热门话题,三维分子结构在蛋白质功能预测、计算机辅助分子设计、合理的药物设计和蛋白质对接等方面有着广泛的应用,使用相关技术研究蛋白质的结构,使人类更加准确地掌握蛋白质结构和功能的知识,是当今生物信息学中最重要的课题之一,将促进生物学、医学、药学等生命科学领域的发展。
随着分子数据库的快速增长,蛋白质的分子研究受益于结构基因组学项目,并以此获取到越来越多的功能仍未知的蛋白质结构。目前的蛋白质数据库已然非常庞大,从数据库中寻找含特定功能和要求的数据需要合适的蛋白质结构编码,以便快速比较。在分子对接过程中,研究者常模拟筛选到的分子与大分子生物受体对接的过程,以估计不同蛋白质的结合性能,这些技术催生了海量的类药物分子数据库。现代化学和药物研究中的三维分子结构数据库相较于原本的分子数据库,三维的分子数据库收集了更加大量的化学知识,比如分子中原子的空间位置,通过包含几个可能的构象来了解分子的灵活性,甚至能进一步分析出分子的生物活性细节,这种特殊形式的扩展,使得三维的数据库应用场景更加广泛,得到的结果更精确。
对三维蛋白质模型检索算法而言,最大的挑战就是形状描述符的设计。由于三维蛋白质表现出丰富的可变性以及蛋白质分子间的互相作用,要求形状描述符在形状的不同转换中保持形式不变,而且要具有对蛋白质变形的不敏感性及易于计算和存储的特点。传统的蛋白质检索方法是基于内容的形状检索,使用蛋白质形状本身作为查询依据,并基于形状的几何拓扑属性的比较,能取得一定的检索效果。但由于三维蛋白质表现出丰富的可变性,而传统方法要求在不同的转换类下,必须保证基本的三维表面形状是不变的,这就使得基于内容的形状检索变得更加复杂,且传统的检索框架对数据的预处理不友好,使得模型冗余检索效率低下,限制了三维蛋白质模型形状检索的发展和应用。
技术实现要素:
本申请实施例提供一种蛋白质检索模型构建方法、检索方法、设备和存储介质,解决相关技术利用蛋白质形状本身作为查询依据时存在的检索效率低下及复杂度高的问题。
本申请解决其技术问题所采用的技术方案如下:
一方面,提供一种蛋白质检索模型构建方法,包括:
对数据集中的所有蛋白质分别生成溶剂排除表面的三角网格,并进行简化和缺陷修复;
用wks算法计算经前一步骤处理后所有三角网格每个顶点的wks特征,选取适量的wks特征,用k均值算法进行聚类,生成第一词典,根据每个蛋白质的三角网格的每个顶点的wks特征和所述第一词典,计算相应蛋白质的第一bof特征;用hks算法计算经前一步骤处理后所有三角网格每个顶点的hks特征,选取适量的hks特征,用k均值算法进行聚类,生成第二词典,根据每个蛋白质的三角网格的每个顶点的hks特征和所述第二词典,计算相应蛋白质的第二bof特征;
将每一个蛋白质的所述第一bof特征和第二bof特征进行归一化处理并拼接,得到每一个蛋白质的第三bof特征;
根据不同蛋白质的第三bof特征的差异程度进行相似度评估;
模型训练,确定所述三角网格模型面大小及所述第一词典和第二词典大小。
优选的,所述三角网格模型面大小为4000,所述第一词典和第二词典大小均为1000。
优选的,所述根据不同蛋白质的第三bof特征的差异程度进行相似度评估,包括:根据归一化后不同蛋白质的所述第三bof特征的l1-范数距离大小评估其形状相似程度。
优选的,所述模型训练,确定三角网格模型面大小及所述第一词典和第二词典大小,包括:
固定所述第一词典和第二词典大小,改变三角网格模型面大小进行训练,根据训练结果确定所述三角网格模型面大小;
依据确定的所述三角网格模型面大小,改变所述第一词典和第二词典大小进行训练,根据训练结果确定所述第一词典和第二词典大小。
另一方面,提供一种蛋白质检索方法,包括:
将待检索蛋白质输入上述方面所述的方法所构建的蛋白质检索模型中,生成溶剂排除表面的三角网格,并进行简化和缺陷修复;
用wks算法计算经前一步骤处理后的所述三角网格每个顶点的wks特征,结合所述蛋白质检索模型的第一词典,计算待检索蛋白质的第一bof特征;用hks算法计算经前一步骤处理后的所述三角网格每个顶点的hks特征,结合所述蛋白质检索模型的第二词典,计算待检索蛋白质的第二bof特征;
将所述第一bof特征和第二bof特征进行归一化处理并拼接,得到待检索蛋白质的第三bof特征;
根据所述第三bof特征与所述蛋白质检索模型中已有蛋白质的第三bof特征的差异程度进行相似度评估,输出与所述待检索蛋白质相似的已知蛋白质的信息。
优选的,相似性评估的方法,包括:根据归一化后待检索蛋白质的所述第三bof特征与所述蛋白质检索模型中已有蛋白质的第三bof特征的l1-范数距离大小评估其形状相似程度。
另一方面,提供一种蛋白质检索模型构建设备,包括至少一个处理器,以及与所述至少一个处理器通信连接的至少一个存储器;
所述至少一个存储器存储有可被所述至少一个处理器执行的指令,所述指令被所述至少一个处理器执行,以使所述至少一个处理器能够执行如上述方面所述的蛋白质检索模型构建方法。
另一方面,提供一种蛋白质检索设备,包括至少一个处理器,以及与所述至少一个处理器通信连接的至少一个存储器;
所述至少一个存储器存储有可被所述至少一个处理器执行的指令,所述指令被所述至少一个处理器执行,以使所述至少一个处理器能够执行如上述方面所述的蛋白质检索方法。
另一方面,提供一种计算机可读存储介质,其上存储有计算机程序,当所述计算机程序被处理执行时,实现如上述方面所述的蛋白质检索模型构建方法,或,如上述方面所述的蛋白质检索方法。
本申请实施例提供的技术方案带来的有益效果至少包括:
对三角网格进行去冗余和去噪声的简化处理,缩短检索时间,提高检索效率;检索算法融合wks与hks两种算法,保持了形状描述符在转换过程中的不变性,加强了形状描述符对蛋白质变形的不敏感性,提高检索精度。实验结果表明,与单独使用wks或hks算法的方法相比,本方法具有更好的检索效果。
附图说明
图1是本申请一个实施例提供的蛋白质检索模型构建方法的流程图;
图2是采用wks算法以及hks算法计算第三bof特征的流程图;
图3是本申请一个实施例提供的蛋白质检索方法的流程图;
图4是模型面为4000、第一词典和第二词典大小均为1000时,本申请一个实施例在molmovdb数据库上的p-r曲线;
图5是模型面为4000、第一词典和第二词典大小均为1000时,本申请一个实施例在shrec2018数据集上的p-r曲线;
图6是本申请一个实施例的设备框图。
具体实施方式
为使本申请的目的、技术方案和优点更加清楚,下面将结合附图对本申请实施方式作进一步地详细描述。
为方便理解,下面对本申请实施例涉及的hks与wks算法进行说明。
热核签名(heatkernelsignature,hks)
hks于2009年提出,是由给定形状的热核导出的标量。它在等距变换下是不变的,还具有鲁棒性以及多尺度特性,反映了模型的固有几何性质,常用于可变形模型的应用,在许多几何应用中得到了显著成果,属于谱形状分析方法,广泛应用于三维分割、分类、结构探索、形状匹配的形状检索。从另一角度来看,hks可以看作黎曼流形度量的描述符,hks特征是由热量在模型表面慢慢传递得到的,构成三维模型所有点的热核的集合,在模型上定义了一个用来测量热量的点在一定的时间内扩散情况的函数。
hks具有等距不变量,包含了物体形状曲面几乎所有的内在信息,更简单的说,hks可以看作曲面的曲率。由于hks是由热扩散过程导出的,故它具有一个时间参数,时间参数是影响hks在某一点的值的邻域大小的度量,基于这些性质,hks被提出来并且形象化对称正定张量场,基本思想是把hks当成曲面的一个特征,可以将hks应用于具有黎曼度量特征的张量场,即对称正定张量场。但hks有两个显著的缺点是缺乏尺度不变性以及对低频信息过于敏感。
hks是基于三维模型上热扩散过程的性质,通过将众所周知的热核限制到时域来获得的,它捕获了所有包含在热核中的信息。根据热扩散理论,设定在三维模型上有热源h0(x),并随时间t在三维模型p表面上进行热量扩散,在一定时刻,形状表面上达到热平衡状态,在这个过程中,定义热核kt(x,y)为t时刻从x点到y点所需的热量,表示热量从一个点到另外一个点的可能性。公式(1)描述了三维模型表面上的温度随时间变化状态。
其中,h(x,t)是三维模型p上时间t对应的热量分布函数,δ是laplace-beltrami(拉普拉斯-贝尔特拉米)算子,这个方程的解是:
h(x,t)=∫kt(x,y)h0(y)dy(2)
对热核进行谱分解:
假设热核在一定的时间域内,可转化为一个简单的形状描述符,hks可表示为:
其中λi和φi分别表示拉普拉斯算子的第i个特征值和相应的特征向量,通过调节时间t改变其描述的是形状的局部特征或者全局特征,三维模型p在不同时间尺度下hks值分布可表示为:
波核签名(wavekernelsignature,wks)
wks于2011年提出,表示在特定位置测量量子力学粒子的平均概率。通过改变粒子的能量,wks编码并从不同的拉普拉斯频率中分离信息。这种尺度分离使得wks适合于各种各样的应用,不管是在理论上还是在定量试验中,wks算法都具有比较强的识别能力。在实际应用中,wks算法对各种类型的扰动具有强鲁棒性,它包括各种受扰动的元素,如噪声、喷射噪声、孔洞、拓扑变化、尺度和局部尺度。
wks源于对控制量子力学粒子在几何表面耗散的肖丁格方程的研究。与hks相比,wks清晰地分离了不同频率的影响,通过理论稳定性分析,确定wks的适当参数,wks对尺度的分离性和对精细尺度信息更好地获取,就算存在强扰动数据,也能正确地检测三维物体的特征。wks解决了hks特征定位差的问题,解决了hks对低频信息的严重敏感问题,不过wks的特征分类能力和形状间匹配精度取决于其参数,除了参数可能影响描述符的特性外,还可能与周围的全局或局部属性的敏感性有关。
根据薛定谔方程,物体表面的每个点都与wks相关联。根据wks对尺度变换的敏感性,引入基于特征值归一化的方法来构造尺度不变波核签名。去除wks中的尺度因子,最后wks定义为:
其中:
σ表示正态分布的方差,λi为拉普拉斯算子第i个特征值对应的特征向量,φi(x)是第i个频率对应的特征向量,ce为正则化wks函数,ei为能量规模参数,ei=log(e),期望log(e)={e1,...em}。
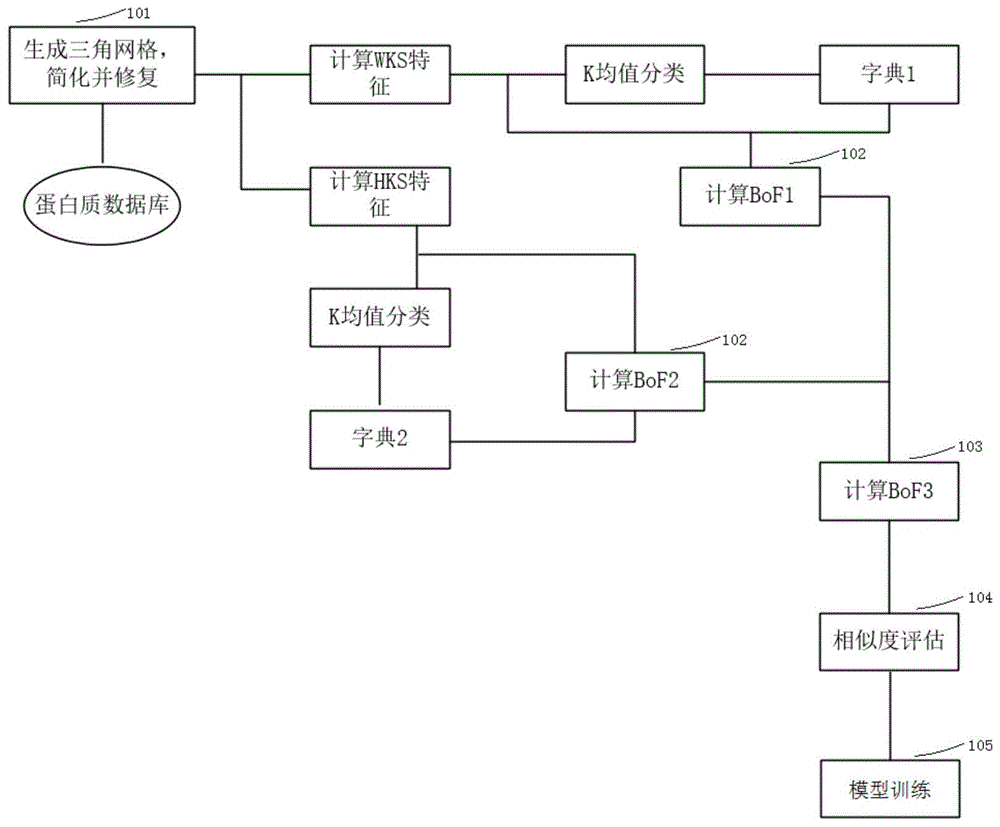
请参考图1,其示出了本申请一个实施例提供的蛋白质检索模型构建方法的流程图,该方法可以包括以下几个步骤:
步骤101,对数据集中的所有蛋白质分别生成溶剂排除表面的三角网格,并进行简化和缺陷修复。
数据集是具有明确分类的蛋白质集合,目前有一些测试数据集合,如fssp(familiesofstructurallysimilarproteins,蛋白质家族数据库)、shrec、molmovdb等。在具体实例中,所使用的数据集为molmovdb和shrec2018数据集,其中,molmovdb:生物大分子运动数据库,由耶鲁大学的生物信息学研究室维护。
在一种可能的实施方式中,三角网格为高分辨率,其顶点数从几十到几十万个,三角面个数可达50万,含有更丰富的分子结构信息。对三角网格进行适当简化,去除高冗余特性和噪声干扰,可缩短算法检索时间,提高模型的运行效率。简化后的三角网格进行缺陷修复,目的是去除重复点、重复面、空洞、自相交、退化以及非流形(non-manifold)等缺陷。
步骤102,用wks算法计算经步骤101处理后所有三角网格每个顶点的wks特征,选取适量的wks特征,用k均值算法进行聚类,每个类心的码字为一个视觉词汇,所有的视觉词汇组成第一词典(图1中的字典1),根据每个蛋白质的三角网格的每个顶点的wks特征和所述第一词典,计算相应蛋白质的第一bof(bagoffeatures,特征包)特征(图1中的bof1)。
用hks算法计算经步骤101处理后所有三角网格每个顶点的hks特征,选取适量的hks特征,用k均值算法进行聚类,生成第二词典(图1中的字典2),根据每个蛋白质的三角网格的每个顶点的hks特征和所述第二词典,计算相应蛋白质的第二bof特征(图1中的bof2)。
步骤103,将每一个蛋白质的第一bof特征和第二bof特征进行归一化处理并拼接,得到每一个蛋白质的第三bof特征(图1中的bof3)。
如图2所示,一个3d蛋白质模型的off格式文件分别用wks算法以及hks算法计算bof,归一化后得到两个长度为m的特征向量,再合并成一个长度为2m的特征向量,即第三bof特征,也是该蛋白质的形状描述符。
步骤104,根据不同蛋白质的第三bof特征的差异程度进行相似度评估。具体的,可根据归一化后不同蛋白质的形状描述符间的l1-范数距离大小评估其形状相似程度,此外还可以采用l2-范数,余弦相似度,曼哈顿距离等进行计算。
对于三维模型x和y,采用两向量的l1距离来度量两个三维模型的相似性,距离越小,表示两个三维模型的形状越相似。相似性可表示为:
d(x,y)=||x-y||1(8)
步骤105,模型训练,确定三角网格模型面大小及第一词典和第二词典大小。采用不同参数设置训练模型,取实验效果最好的一种参数组合,作为最终参数。
比如,可以采用以下方式进行训练:
f1:固定第一词典和第二词典大小均为500;
f2:分别采用2000,4000,6000,8000大小的三角网格进行训练;
f3:根据训练结果确定最佳三角网格模型面大小为4000;
f4:固定三角网格模型面大小为4000;
f5:第一词典和第二词典取相同大小,分别采用100,500,1000,1500的词典进行训练;
f6:根据训练结果确定最佳词典大小为1000,即第一词典和第二词典大小均为1000。
本实施例的模型训练过程,第一词典和第二词典大小相同,当然,两个词典大小也可以不同。
请参考图3,其示出了本申请一个实施例提供的蛋白质检索方法的流程图。该方法应用于采用前述方法构建的蛋白质检索模型中,对于本方法中未披露的细节,请参照本申请蛋白质检索模型构建方法实施例。本方法可以包括以下几个步骤:
步骤201,将待检索蛋白质输入该蛋白质检索模型中,生成溶剂排除表面的三角网格,进行简化,去除高冗余信息和噪声信息,然后进行缺陷修复,去除重复点、重复面、空洞、自相交、退化以及非流形等缺陷。
步骤202,用wks算法计算经步骤201处理后的三角网格每个顶点的wks特征,结合蛋白质检索模型的第一词典(图3中的字典1),计算待检索蛋白质的第一bof特征(图3中的bof1);用hks算法计算经步骤201处理后的三角网格每个顶点的hks特征,结合蛋白质检索模型的第二词典(图3中的字典2),计算待检索蛋白质的第二bof特征(图3中的bof2)。
步骤203,将该第一bof特征和第二bof特征进行归一化处理并拼接,得到第三bof特征(图3中的bof3),作为待检索蛋白质的形状描述符。
步骤204,根据该第三bof特征与蛋白质检索模型中已有蛋白质的第三bof特征的差异程度进行相似度评估,评估方法与输出与所述待检索蛋白质相似的已知蛋白质的信息。
以不同蛋白质第三bof特征的l1-范数距离大小来评估其形状相似程度,取模型面为4000,词典大小1000,按上述实施例的检索方法对模型的检索效果进行评价。广泛应用的评价标准有p-r曲线(p为查准率,r为查全率)、最近邻方法(nearestneighbor,nn)、第1层级(first-tier,ft)、第2层级(second-tier,st)、e度量(e-measure,e)、累积加权值(discountedcumulativegain,dcg)(p.shilane;p.min;m.kazhdan;t.funkhouser.theprincetonshapebenchmark.proceedingsshapemodelingapplications,2004,167-178.)。
本实施例在molmovdb数据库的p-r曲线如图4所示,图中混合谱算法代表本实施例,其余评价指标见表1。在本实施例的基础上,仅采用wks算法或hks算法进行计算,相应的检索性能比较如表1所示。
表1
本实施例在shrec2018数据集的p-r曲线如图5所示,图中混合谱算法代表本实施例,在本实施例的基础上,仅采用wks算法或hks算法进行计算,相应的检索性能比较见表2。
表2
由以上结果可知,本实施例在各数据集上的检索性能均优于单独的wks算法或hks算法。
上述方面所述的蛋白质检索模型构建方法和蛋白质检索方法可以在同一设备上实现,该设备可以是计算机或服务器,包括至少一个处理器301和至少一个存储器302,图6仅以一个处理器301和一个存储器302为例。存储器302存储有软件程序、指令以及数据集等信息,处理器通过运行存储在存储器302中的软件程序、指令以及调用数据集中的数据,从而执行前述的蛋白质检索模型构建方法或蛋白质检索方法。
本申请一个实施例提供的一种计算机可读存储介质,所述计算机可读存储介质存储有计算机程序,该计算机程序被一个或多个处理器执行,例如,被图6中的一个处理器301执行,可使得上述一个或多个处理器执行上述方法实施例中的蛋白质检索模型构建方法,例如,执行以上描述的图1中的方法步骤101至105;或者,执行上述方法实施例中的蛋白质检索方法,例如,执行以上描述的图3中的方法步骤201至204。
以上实施例是对本申请的解释,但是,本申请并不局限于上述实施方式中的具体细节,本领域的技术人员在本申请的技术构思范围内进行的多种等同替代或简单变型方式,均应属于本申请的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!