基于光调节的聚乙二醇化多肽前药合成工艺及其在制备抗菌药物中的用途的制作方法
本发明属药物合成领域,具体涉及光敏感复合物cgs-mpeg及cin-mpeg的合成工艺及其在制备抗菌药物中的用途。
背景技术:
天然抗菌肽(antimicrobialpeptides,amps)是生物体经诱导产生的具有抗菌活性的一类小分子多肽,来源广泛,其分子量小,大约在1~6kd之间,耐热、耐酸性强,水溶性好,显示出高效的杀菌能力和广谱的抗菌活性,包括革兰氏阳性菌、革兰氏阴性菌、真菌、寄生虫、病毒和肿瘤细胞。抗菌肽作用机制独特,主要是通过破坏细菌细胞膜造成胞质流失而诱导细菌死亡,该作用机制最大的特点在于不容易引起细菌耐药性。在当前耐药菌形势严峻的情况下,抗菌肽不失为一种具有广泛使用前景的抗菌药物。然而,天然抗菌肽往往具有较强的溶血副作用,且对酶解代谢稳定性不高,这些缺点严重影响了抗菌肽的临床使用。为此,在不影响抗菌肽生物活性的情况下,改善其溶血及细胞毒活性,并提高代谢稳定性以适应临床需求是目前抗菌肽药物研发领域的热点课题。
本发明利用前药原理,以gramicidins(gs)和indolicidin(in)为例,成功地开发出一条基于光引发的抗菌肽前药的合成路线,并对其进行体外研究。在对抗菌肽聚乙二醇化修饰后,多肽前药的溶血和细胞毒活性明显降低,且代谢稳定性大幅提高。在经过紫外光照射之后,多肽前药可重新释放出具有活性的抗菌肽分子。由于未经紫外照射的多肽前药未显示出抗菌活性,具有明显的专一性和特殊性。为此,该项合成技术可有效地进行靶向治疗,且通过光调节活性药物的释放浓度及作用部位,这对于后期开发天然抗菌肽作为临床抗癌药、抗菌药具有重要意义。
技术实现要素:
本发明的目的是合成具有感光活性的天然抗菌肽(antimicrobialpeptides,amps),具体涉及gramicidins(gs)和indolicidin(in)感光化合物的合成制备方法和在抑菌活性中的用途。
本发明通过药物化学合成方法,将gramicidins(gs)中的鸟氨酸残基与光敏感基团2-nitrobenzyl(nb)进行连接,indolicidin(in)中的赖氨酸与光敏感基团2-nitrobenzyl(nb)进行连接,并连接不同分子量的单甲氧基聚乙二醇(mpeg),使其溶血及细胞毒活性消失。虽然抑菌活性也受到抑制,但通过紫外照射后,其抗菌肽gs和in以原型释放,在光照射部位重新显示出抗菌活性.本发明不仅适用于环状抗菌肽gramicidins也适用于链状抗菌肽indolicidin。
本发明所述的由gramicidins(gs)合成的光敏感复合物cgs-mpeg的结构特征描述如下:
1.cgs-mpeg复合物的光解动力学曲线:高效液相色谱hplc检测cgs-mpeg复合物在uv光照下(365nm,172mw/cm2)的光解曲线(0-160min);随着uv照射时间延长,cgs-mpeg逐渐分解,而gs原型则缓慢增加,说明该复合物分解与uv照射的时间呈正相关,见图2和图3;
2.cgs-mpeg复合物质谱图,经过160min的照射,从质谱中明显出现gs和nb-mpeg的质谱峰.验证了cgs-mpeg的组成,见图4;
cgs-mpeg活性评价如下:
1.通过体外溶血实验发现cgs-mpeg溶血活性显著降低,且在<100um的浓度范围内,对溶血副作用是基本可以忽略,见图5;
2.通过细胞毒活性实验发现cgs-mpeg浓度在1-25um范围内,不显示细胞毒活性,而其原型gs在浓度>5um时具有显著的细胞毒活性,说明cgs-mpeg对溶血副作用是基本可以忽略,见图6及图7;
3.通过抑菌实验发现cgs-mpeg经uv照射后,会分解出gs原型,产生明显的抑菌活性,而未被照射的地方,细菌正常生长,因此呈现出与照射范围类似的菌圈,见图8。
本发明所述的由indolicidin合成的光敏感复合物cin-mpeg的结构特征描述如下:
1.cin-mpeg复合物的光解动力学曲线:高效液相色谱hplc检测cin-mpeg复合物在uv光照下(365nm,172mw/cm2)的光解曲线(0-60min);随着uv时间的照射时间延长,cin-mpeg逐渐分解,而indolicidin原型则缓慢增加,说明该复合物分解与uv照射的时间呈正相关,见图9和图10;
2.cin-mpeg复合物质谱图,经过60min的照射,从质谱中明显出现indolicidin和nb-mpeg的质谱峰.验证了cin-mpeg的组成,见图11;
cin-mpeg活性评价如下:
1.通过体外溶血实验发现cin-mpeg溶血活性显著降低,且在<100um的浓度范围内,对溶血副作用是基本可以忽略,见图12。
2.通过细胞毒活性实验发现cin-mpeg浓度在1-25um范围内,不显示细胞毒活性,而其原型gs在浓度>25um时具有显著的细胞毒活性,说明cgs-mpeg对溶血副作用是基本可以忽略,见图13及图14。
附图说明
图1为光敏感鸟氨酸和赖氨酸的合成路线,以及叠氮化mpeg的合成路线;
图2为cgs-mpeg2000光解动力学hplc分析图;
图3为cgs-mpeg2000光照时间-峰面积图;
图4为cgs-mpeg2000光解产物质谱图;
图5为cgs-mpeg复合物与gs溶血副作用比较;
图6为cgs-mpeg2000与gs细胞毒性比较(10%胎牛血清培养条件);
图7为cgs-mpeg2000与gs细胞毒性比较(1%胎牛血清培养条件);
图8为细菌培养光照造型实验,接受光照的区域没有细菌生长,反之则细菌正常生长;
图9为cin-mpeg2000光解动力学hplc分析图;
图10为cin-mpeg2000光照时间-峰面积图;
图11为cin-mpeg2000光解产物质谱图;
图12为cin-mpeg2000与in溶血副作用比较;
图13为cin-mpeg2000与in细胞毒性比较(10%胎牛血清培养条件);
图14为cin-mpeg2000与in细胞毒性比较(1%胎牛血清培养条件);
图15为cgs-mpeg2000核磁氢谱图;
图16为cgs-mpeg2000质谱图;
图17为gs核磁氢谱图;
图18为gs质谱图;
图19为cin-mpeg2000质谱图。
具体实施方式
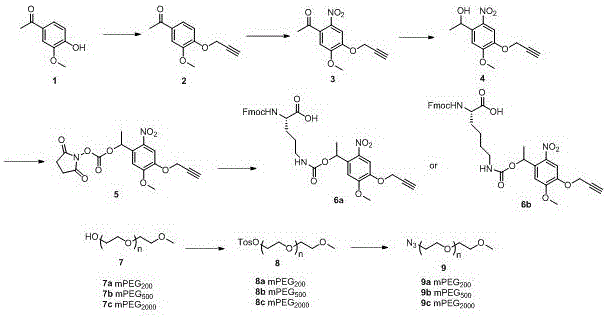
以4'-羟基-3'-甲氧基苯乙酮为起始原料制备光敏感基团2-nitrobenzyl(nb)。首先第一步用炔丙基溴取代酚羟基,以避免多余的保护和脱保护的步骤,第二步通过加入适量kno3和tfa对苯环上2号位进行硝基取代,第三步将羰基还原并用n,n'-琥珀酰亚胺基碳酸酯对其进行保护以制得光敏感基团2-nitrobenzyl(nb)。
通过在侧链氨基位置引入光敏感基团2-nitrobenzyl(nb)来预先制备光敏感的鸟氨酸和赖氨酸。通过固相多肽合成制备得到含光敏感基团的gs和in。多肽与peg的连接通过“点击”反应的策略,mpeg通过叠氮取代,与光敏感基团的炔烃发生‘点击’反应从而将多肽与mpeg进行连接,制备得到cgs-mpeg和cin-mpeg。
反应流程图如下:
合成化合物2:将化合物1(10g,60.2mmol)溶解于150ml无水乙醇中,向其加入炔丙基溴(6.8ml,90.3mmol)和无水碳酸钾(16.6g,120.2mmol),在78℃下过夜搅拌16h;反应完毕,旋干反应液,剩余物溶于300ml乙酸乙酯中,并用1n盐酸(300ml)、饱和碳酸氢钠水溶液(300ml)、饱和食盐水(300ml)洗涤,无水硫酸镁干燥有机层,过滤并减压浓缩;通过色谱法(sio2,乙酸乙酯/石油醚,3/7,v/v)纯化粗产物,得到11.2g化合物2,收率为91%;esi-ms:calcdforc12h13o3([m+h]+)205.09,found205.11.c12h12nao3([m+na]+)227.07,found227.13。
合成化合物3:在0℃下向硝酸钾(1.338g,13.2mmol)的三氟乙酸(21.8ml,294mmol)溶液中滴加化合物2(3g,14.7mmol)的三氟乙酸(21.8ml,294mmol)溶液;在室温下搅拌16h;反应毕,旋干反应液;剩余物溶于二氯甲烷(200ml)中,并用饱和碳酸氢钠水溶液(200ml)、饱和氯化钠水溶液(200ml)洗涤;无水硫酸镁干燥有机层,过滤并减压浓缩;通过色谱法(sio2,乙酸乙酯/石油醚,3/7,v/v)纯化粗产物,得到1.6g化合物3,收率为49%;esi-ms:calcdforc12h12no5([m+h]+)250.07,found250.29;c12h15n2o5([m+nh4]+)267.10,found267.11;c12h11nnao5([m+na]+)272.05,found272.26。
合成化合物4:在0℃下向化合物3(1.5g,6.02mmol)的甲醇/1,4二氧六环(15ml/15ml,v/v)中溶液中加入硼氢化钠(456mg,12.05mmol);室温下搅拌反应2h。反应毕,旋干反应液;剩余物溶于二氯甲烷(150ml)中,并用1n盐酸水溶液,纯水洗涤。无水硫酸镁干燥有机层,过滤并减压浓缩。通过色谱法(sio2,乙酸乙酯/石油醚,3/7,v/v)纯化粗产物,得到1.45g化合物4,收率为96%;esi-ms:calcdforc12h12no5([m-h]-)250.07,found250.14。
合成化合物5:将化合物4(1.39g,5.54mmol)溶解于40ml乙腈中,加入三乙胺(3ml,22.15mmol)和n,n'-二琥珀酰亚胺基碳酸酯(4.26g,16.61mmol);在室温下搅拌反应5h;反应毕,旋干反应液;剩余物溶于二氯甲烷(100ml)中,并用1n盐酸水溶液(100ml)、饱和碳酸氢钠水溶液(100ml)、饱和氯化钠(100ml)水溶液洗涤,无水硫酸镁干燥有机层,过滤并减压浓缩;通过色谱法(sio2,乙酸乙酯/石油醚,4/6,v/v)纯化粗产物,得到2.09g化合物5,收率为96%;esi-ms:calcdforc17h20n3o9([m+nh4]+)410.12,found410.08;c17h16n2nao9([m+na]+)415.07,found414.93。
合成化合物6b:将化合物5(420mg,1.07mmol)溶解二氯甲烷/乙腈(10ml/10ml,v/v)中,向其加入n-芴甲氧羰基-l-鸟氨酸(417mg,1.18mmol)和n,n-二异丙基乙胺(0.53ml,3.21mmol);在室温下搅拌反应12h,反应毕,旋干反应液;剩余物溶于二氯甲烷(100ml)中,并用1n盐酸水溶液,纯水洗涤;无水硫酸镁干燥有机层,过滤并减压浓缩;通过色谱法(sio2,甲醇/二氯甲烷,3/97,v/v)纯化粗产物,得到570mg化合物6a,收率为84%;esi-ms:calcdforc33h32n3o10([m-h]-)630.21,found630.12.
合成化合物6b:将化合物5(420mg,1.07mmol)溶解二氯甲烷/乙腈(10ml/10ml,v/v)中,向其加入n-芴甲氧羰基-l-鸟氨酸(420mg,1.14mmol)和n,n-二异丙基乙胺(0.53ml,3.21mmol);在室温下搅拌反应12h,反应毕,旋干反应液;剩余物溶于二氯甲烷(100ml)中,并用1n盐酸水溶液,纯水洗涤;无水硫酸镁干燥有机层,过滤并减压浓缩;通过色谱法(sio2,甲醇/二氯甲烷,3/97,v/v)纯化粗产物,得到570mg化合物6a,收率为75%;esi-ms:calcdforc33h32n3o10c34h39n4o10([m+nh4]+)663.27;found:662.94.
化合物8a、8b、8c的合成分别以不同长度mpeg为底物,反应条件一致,以化合物8a合成为例:将7a(1.5g,7.2mmol)溶解于甲苯(70ml)和二氯甲烷(35ml)中;向其中加入对甲苯磺酰氯(11.44g,72mmol)和三乙胺(9.7ml,72mmol);在室温下搅拌反应16h;反应毕,滤除不溶物,减压浓缩滤液。通过色谱法(sio2,甲醇/二氯甲烷,3/97,v/v)纯化粗产物,得到2.43g化合物8a,收率为93.2%;当以化合物7b和7c作为原料时,化合物8b和8c的收率分别为68%和54%。
化合物9a-9c分别以8a-8c为底物,反应条件一致,以化合物9a合成为例:将化合物8a(1.2g,3.31mmol)溶解于二甲基甲酰胺(10ml)中,向其加入叠氮钠(323mg,4.97mmol),在90℃下搅拌反应12h,反应毕,蒸除二甲基甲酰胺溶剂;通过色谱法(sio2,甲醇/二氯甲烷,2/98,v/v)纯化粗产物,得到691mg化合物9a,收率为83%;当以化合物8b和8c作为原料时,化合物9b及9c收率分别为78%和71%。
合成化合物11:多肽合成采用固-液相结合的合成方式,固相合成主要通过cemdiscoverbio半自动微波多肽合成仪完成;合成以n-叔丁芴甲氧羰基-l-脯氨酸-2-氯三苯甲基氯树脂(1%交联,100-200目,0.476mmol/g)作为固相载体,将n-叔丁芴甲氧羰基-l-脯氨酸-2-氯三苯甲基氯树脂(210mg,0.1mmol)放置反应器中进行多肽合成;
(a)肽链延长,循环步骤如下:
(1)fmoc脱保护:加入20%哌啶的二甲基甲酰胺溶液(4ml),反应温度为50℃,微波功率30w,反应时间为210s;(2)肽偶联:加入fmoc-aa-oh(5当量),o-苯并三氮唑-四甲基脲六氟磷酸酯(5当量)和n,n-二异丙基乙胺(在dmf中为0.3m),无水二甲基甲酰胺(4ml);反应温度为50℃,微波功率30w,反应时间为600s;(3)洗涤:每次脱保护或偶联后,用二甲基甲酰胺洗涤树脂三次(4ml/次)。
合成化合物12:
(b)切割树脂:将化合物11转移至多肽合成管中,用1%三氟乙酸的二氯甲烷溶液切割4次(8ml/次),反应时间为10min;切割后,将产物溶液与甲苯(1/1,v/v)共蒸发,得到固体侧链保护的粗线性肽;
(c)多肽环化:向六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(5.0当量)和1-羟基苯并三唑(5.0当量)的无水二甲基甲酰胺溶液(2ml/mg)中加入n,n-二异丙基乙胺(15.0当量);在1小时内逐滴添加溶解于无水二甲基甲酰胺(10ml)中的线性肽;将溶液在室温搅拌过夜后,减压除去溶剂;
(d)脱出侧链boc保护基:将环化的肽溶解于三氟乙酸/水(10mg/ml,9:1)中;将溶液在室温搅拌1h;减压浓缩反应液至剩余约1ml溶剂,将等体积(0.5ml)的三氟乙酸溶液等分试样倒入两个15ml离心管中;然后将冷的乙醚沿壁缓慢加入到离心管中;立即观察到沉淀,并在冰浴中孵育15分钟;离心(4000rpm,10分钟)沉淀出沉淀物,并在除去乙醚上清液后转移至圆底烧瓶中;通过半制备性rp-hplc纯化并冻干,得到36mg白色粉末状化合物12,收率为25%。
cgs-mpeg复合物合成分别以不同长度叠氮化mpeg为底物,反应条件一致,以cgs-mpeg2000合成为例:将化合物12(15mg,0.011mmol)溶解于水/叔丁醇(4ml/4ml,v/v)中,向其加入n3-peg2000(23.7mg,0.012mmol)五水硫酸铜(3.96mg,0.016mmol)和抗坏血酸钠(5.24mg,0.026mmol);在50℃下搅拌反应20min,反应毕,旋干反应液;通过制备液相进行提纯,得到11mgcgs-mpeg2000,收率为25%;制备cgs-mpeg200和cgs-mpeg500收率分别为43%和36%。
合成化合物14:多肽合成采用固-液相结合的合成方式,固相合成主要通过cemdiscoverbio半自动微波多肽合成仪完成;合成以n-叔丁芴甲氧羰基-l-脯氨酸-2-氯三苯甲基氯树脂(1%交联,100-200目,0.476mmol/g)作为固相载体,将n-叔丁芴甲氧羰基-l-脯氨酸-2-氯三苯甲基氯树脂(210mg,0.1mmol)放置反应器中进行多肽合成;
(a)肽链延长,循环步骤如下:
(1)fmoc脱保护:加入20%哌啶的二甲基甲酰胺溶液(4ml),反应温度为50℃,微波功率30w,反应时间为210s;(2)肽偶联:加入fmoc-aa-oh(5当量),o-苯并三氮唑-四甲基脲六氟磷酸酯(5当量)和n,n-二异丙基乙胺(在dmf中为0.3m),无水二甲基甲酰胺(4ml);反应温度为50℃,微波功率30w,反应时间为600s;(3)洗涤:每次脱保护或偶联后,用二甲基甲酰胺洗涤树脂三次(4ml/次)。
合成化合物15:
(b)切割树脂及侧链保护基:将化合物14转移至多肽合成管中,用95%三氟乙酸的水溶液切割2次(8ml/次),反应时间为30min;减压浓缩切割液至剩余约1ml溶剂,将等体积(0.5ml)的三氟乙酸溶液等分试样倒入两个15ml离心管中;然后将冷的乙醚沿壁缓慢加入到离心管中;立即观察到沉淀,并在冰浴中孵育15分钟;离心(4000rpm,10分钟)沉淀出沉淀物,并在除去乙醚上清液后转移至圆底烧瓶中;通过半制备性rp-hplc纯化并冻干,得到19mg白色粉末状化合物15,收率为8.7%。
合成cin-mpeg2000:将化合物15(17mg,0.0078mmol)溶解于水/叔丁醇(4ml/4ml,v/v)中,向其加入n3-peg2000(17.5mg,0.0086mmol)五水硫酸铜(2.92mg,0.0117mmol)和抗坏血酸钠(3.70mg,0.0187mmol);在50℃下搅拌反应20min,反应毕,旋干反应液;通过制备液相进行提纯,得到9mgcin-mpeg2000,收率为28%。
- 还没有人留言评论。精彩留言会获得点赞!