使用胶态铝硅酸盐和新型结构导向剂制备CHA型分子筛的方法与流程
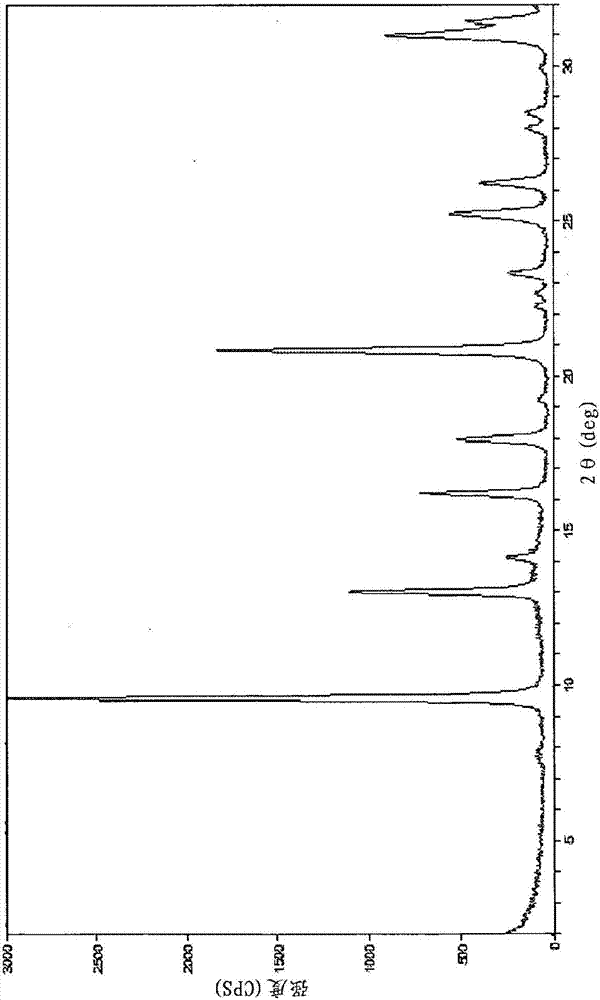
本发明涉及使用胶态铝硅酸盐组合物和选自N-环己基-N-甲基吡咯烷鎓、N-甲基-N-(3-甲基环己基)吡咯烷鎓、N-乙基-N-(3-甲基环己基)吡咯烷鎓及其混合物的阳离子型结构导向剂制备CHA型分子筛的方法。发明背景分子筛是在商业上重要的一类结晶材料。它们具有通过不同的X射线衍射图案证实的具有有序孔隙结构的不同晶体结构。该晶体结构限定了作为不同物类的特征的空腔与孔隙。由国际沸石协会(IZA)识别为具有结构代码CHA的分子筛是已知的。例如,称为SSZ-13的分子筛是已知的结晶CHA材料。其公开在1985年10月1日授予Zones的美国专利号4,544,538中。在该专利中,在作为结构导向剂(SDA)的N-烷基-3-喹核醇阳离子、N,N,N-三烷基-1-金刚烷铵阳离子和/或N,N,N-三烷基-2-外氨基降冰片烷阳离子的存在下制备SSZ-13分子筛。2007年12月13日公开的授予Cao等人的美国公开号2007-0286798公开了使用各种SDA,包括N,N,N-三甲基-2-金刚烷铵阳离子制备CHA型分子筛。但是,可用于制造CHA材料的已知SDA是复杂的,并且通常不能以商业规模生产CHA材料所需的量获得。此外,始终需要降低反应混合物中已知CHASDA的浓度至绝对最小值,或用更廉价、更简单和/或减少形成产物所需时间的SDA完全替代它们。现在已经发现,CHA型分子筛可以使用胶态铝硅酸盐在至少一种下文中描述的新型结构导向剂的存在下制备。发明概述根据本发明,提供了通过在结晶条件下使以下成分接触来制备CHA型分子筛的方法:(1)胶态铝硅酸盐组合物;(2)至少一种下文中结构(1)至(3)所代表的新型结构导向剂;(3)选自周期表第1和2组的元素的至少一种来源;和(4)氢氧根离子。本发明还包括通过以下步骤制备CHA型分子筛的方法:(a)制备含有以下成分的反应混合物:(1)胶态铝硅酸盐组合物;(2)至少一种下文中结构(1)至(3)所代表的新型结构导向剂;(3)选自周期表第1和2组的元素的至少一种来源;(4)氢氧根离子;和(5)水;以及(b)对该反应混合物施以足以形成CHA型分子筛的晶体的结晶条件。当形成的分子筛是中间体材料时,本发明的方法包括在结晶后的进一步处理以获得目标分子筛(例如通过合成后的杂原子晶格置换或酸浸出)。本发明还提供了CHA型分子筛,合成后原样且在无水状态下的该分子筛具有以摩尔比计的如下组成:其中:(1)M选自周期表第1和2族的元素;和(2)Q是至少一种下文中结构(1)至(3)所代表的新型结构导向剂。附图概述图1显示了按照本发明的实施例6制备的制得后原样的铝硅酸盐CHA分子筛的粉末X射线衍射(XRD)图谱。图2显示了按照本发明的实施例6制备的煅烧的铝硅酸盐CHA分子筛的粉末XRD图谱。图3显示了按照本发明的实施例8制备的制得后原样的铝硅酸盐CHA分子筛的粉末XRD图谱。图4显示了按照本发明的实施例8制备的煅烧的铝硅酸盐CHA分子筛的粉末XRD图谱。图5显示了按照本发明的实施例10制备的制得后原样的铝硅酸盐CHA分子筛的粉末XRD图谱。图6显示了按照本发明的实施例10制备的煅烧的铝硅酸盐CHA分子筛的粉末XRD图谱。发明详述引言术语“周期表”指的是日期为2007年6月22日的IUPAC元素周期表版本,该周期表族的编号方案如ChemicalandEngineeringNews,63(5),27(1985)中所述。术语“分子筛”包括(a)中间体和(b)通过(1)直接合成或(2)结晶后处理(二次合成)制得的最终或目标分子筛与沸石。二次合成技术能够通过杂原子晶格置换或其它技术由中间体材料合成目标材料。例如,可以由中间体硼硅酸盐通过结晶后Al对B的杂原子晶格置换来合成铝硅酸盐。此类技术是已知的,例如描述在2004年9月14日授予C.Y.Chen和StaceyZones的美国专利号6,790,433中。术语“CHA型分子筛”包括指定为国际沸石协会骨架代码CHA的所有分子筛及其同型物(isotype),如AtlasofZeoliteFrameworkTypes,编者Ch.Baerlocher,L.B.McCusker和D.H.Olson,Elsevier,第6修订版,2007中所述。该AtlasofZeoliteFrameworkTypes尤其将两种不同名称的材料分类为具有相同的拓扑结构:SSZ-13和SSZ-62。本领域技术人员将理解,按照本文中描述的方法制得的CHA型分子筛材料可以含有杂质,如非晶材料;具有非CHA骨架拓扑结构的单元晶胞(例如MFI、MTW);和/或其它杂质(例如重金属和/或有机烃类)。如果允许的话,本申请中引用的所有出版物、专利和专利申请经此引用全文并入本文;其程度使得此类公开不与本发明不一致。除非另行指明,描述可从中选择单个组分或组分的混合物的元素、材料或其它组分的类别意在包括列举的组分及其混合物的所有可能的子类别组合。同样,“包括”及其变体意在是非限制性的,使得提及列表中的项目并非想要排除也可用于本发明的材料、组合物与方法的其它类似项目。本发明涉及使用胶态铝硅酸盐和环状含氮阳离子结构导向剂(SDA)制造CHA型分子筛的方法,所述SDA选自结构(1)至(3)所表示的阳离子,及其混合物:N-环己基-N-甲基吡咯烷鎓阳离子N-甲基-N-(3-甲基环己基)吡咯烷鎓阳离子N-乙基-N-(3-甲基环己基)吡咯烷鎓阳离子反应混合物通常,通过以下步骤制备CHA型分子筛:(a)制备含有以下成分的反应混合物:(1)胶态铝硅酸盐组合物;(2)至少一种本文中结构(1)至(3)所代表的新型结构导向剂;(3)选自周期表第1和2族的元素的至少一种来源;(4)氢氧根离子;和(5)水;以及(b)对该反应混合物施以足以形成CHA型分子筛的晶体的结晶条件。当形成的分子筛是中间体材料时,本发明的方法包括通过合成后技术如杂原子晶格置换和酸浸出来合成目标分子筛的进一步的步骤。在下表1中确定了由此形成CHA型分子筛的反应混合物的组成,以摩尔比计:表1其中组成变量M和Q如上文中所述。可用于本文中所述的方法并通常用于制造分子筛的胶态铝硅酸盐组合物在本领域是公知的,并可购自供应商如Nalco。如上文中所述,对本文中所述的各实施方案,可以使用选自周期表第1和2组的元素的至少一种来源(在本文中称为M)形成该反应混合物。在一个子实施方案中,使用选自周期表第1族的元素的来源形成反应混合物。在另一子实施方案中,使用钠(Na)源形成该反应混合物。对结晶过程无害的任何含M化合物是合适的。此类第1和2族元素的来源包括其氧化物、氢氧化物、硝酸盐、硫酸盐、卤化物、草酸盐、柠檬酸盐和乙酸盐。SDA阳离子通常与阴离子(X-)相关联,其可以是对形成沸石无害的任何阴离子。代表性阴离子包括选自周期表第17族的元素(例如氟离子、氯离子、溴离子和碘离子)、氢氧根、乙酸根、硫酸根、四氟硼酸根、羧酸根等等。反应混合物可以批量或连续制备。本文中描述的分子筛的晶体尺寸、形貌和结晶时间可以随反应混合物的性质和结晶条件不等。结晶和合成后处理在实践中,通过以下步骤制备该分子筛:(a)制备如上文中所述的反应混合物;和(b)将所述反应混合物保持在足以形成该分子筛的结晶条件下。(参见HarryRobson,VerifiedSynthesesofZeoliticMaterials,第2修订版,Elsevier,Amsterdam(2001))。该反应混合物保持在提高的温度下直到形成分子筛。水热晶化通常在压力下且通常在高压釜中进行,以使得该反应混合物在130℃至200℃的温度下经受自生压力一至六天。在晶化步骤过程中可以对该反应混合物施以温和搅拌或搅动。本领域技术人员将理解,本文中所述的分子筛可以含有杂质,如非晶材料、具有与该分子筛不一致的骨架拓扑结构的单元晶胞、和/或其它杂质(例如有机烃类)。在水热晶化步骤过程中,可以令该分子筛晶体从反应混合物中自发成核。使用该分子筛的晶体作为晶种可以有利地减少发生完全晶化所需的时间。此外,引晶可以通过促进经任何不需要的阶段的成核和/或分子筛形成来提高获得的产物的纯度。当用作晶种时,以反应混合物中所用硅源的重量的1%至10%的量加入晶种。一旦形成分子筛晶体,通过标准机械分离技术如过滤从该反应混合物中分离固体产物。将该晶体水洗并随后干燥以获得合成后原样的分子筛晶体。可以在大气压下或在真空下进行该干燥步骤。该分子筛可以依合成后原样使用,但通常进行热处理(煅烧)。术语“合成后原样”指的是处于结晶后、去除SDA之前的形式的分子筛。该SDA可以通过热处理(例如煅烧)除去,优选在氧化性气氛(例如空气、氧分压大于0kPa的气体)中在足以从分子筛中除去该SDA的容易由本领域技术人员确定的温度下。该SDA还可以通过如2005年11月1日授予Navrotsky和Parikh的美国专利号6,960,327中所述的光催化技术(例如在足以选择性地从分子筛中除去有机化合物的条件下将含有SDA的分子筛产品暴露于波长短于可见光的电磁辐射或光)除去。该分子筛可以随后在水蒸气、空气或惰性气体中在约200℃至约800℃的温度下煅烧1至48小时的时间或更久。通常合意的是通过离子交换或其它已知方法除去骨架外阳离子(例如H+)并用氢、铵或任何所需金属离子取代它。当形成的分子筛是中间体材料时,可以使用合成后技术如杂原子晶格置换技术来获取目标分子筛。目标分子筛(例如硅酸盐SSZ-13)还可以通过已知技术如酸浸出从晶格中除去杂原子来实现。由本发明的方法制得的分子筛可以成形为多种物理形状。一般来说,该分子筛可以是粉末、颗粒或模塑产品如具有足以通过2目(泰勒)筛并保留在400目(泰勒)筛上的粒度的挤出物的形式。在其中模塑该催化剂的情况下,如通过使用粘合剂的挤出法,该分子筛可以在干燥前挤出,或干燥或部分干燥并随后挤出。该分子筛可以与耐受有机转化过程中采用的温度和其它条件的其它材料复合。此类基质材料包括活性和惰性材料,以及合成或天然存在的沸石以及无机材料如粘土、二氧化硅和金属氧化物。此类材料和可以使用它们的方式的实例公开在1990年5月20日授予Zones等人的美国专利号4,910,006和1994年5月31日授予Nakagawa的美国专利号5,316,753中。分子筛的表征通过本发明的方法制得的CHA分子筛依合成后原样且在无水状态下具有如表2中所述的组成(以摩尔比计),其中组成变量M和Q如上文中所述:表2通过本发明的方法合成的CHA型分子筛通过它们的X射线衍射图谱(XRD)来表征。代表CHA型分子筛的X射线衍射图谱可以参考国际沸石协会的M.M.J.Treacy等人,“CollectionofSimulatedXRDPowderPatternsforZeolites”,第五修订版,2007。衍射图谱中的微小变化可能来自于晶格常数改变造成的特定样品的骨架物类的摩尔比的变化。此外,足够小的晶体会影响峰的形状和强度,导致显著的峰宽化。衍射图谱中的微小变化还可能来自于制备中使用的有机化合物的改变和来自于样品与样品之间Si/Al摩尔比的改变。煅烧也可能导致X射线衍射图谱中的微小偏移。尽管存在这些微小的扰动,基本的晶格结构保持不变。本文中呈现的粉末X射线衍射图谱通过标准技术来收集。辐射是CuK-α辐射。作为2θ(其中θ是布拉格角)的函数的峰高度和位置从峰的相对强度读取,并且可以计算对应于记录的谱线的晶面间距d(以埃为单位)。实施例下面的实施例展示但不限制本发明。实施例1合成环己基吡咯烷如下文图式1中所示程序所述制备环己基吡咯烷。在配备有机械搅拌器和加热套的三颈反应烧瓶中,将2摩尔当量的吡咯烷与干燥的环己烷中的1摩尔当量环己酮混合以制造相对于环己酮的1M溶液。向该混合物中加入2摩尔当量的无水MgSO4(作为脱水剂)。所得混合物回流94小时。反应进程通过NMR分析来监控。在加热96小时后,反应完成,将反应混合物冷却,过滤并在旋转蒸发仪上浓缩以除去过量的吡咯烷和环己酮。获得的油溶解在环己烷中并在活性碳上10%钯(相对于制得的环己烯基吡咯烷为10摩尔%)的存在下在Parr氢化设备上在60psi氢压力下氢化。令反应轻轻摇晃过夜。一旦氢化完成,该反应混合物通过烧结玻璃漏斗过滤并将滤液在旋转蒸发仪上浓缩以除去溶剂(环己烷)。以91%的产率获得所需的环己烷基吡咯烷(cyclohexanylpyrrolidine)。图式1在一些情况下,当一些未反应的环己酮仍在反应混合物中时,氢化后的产物通过向混合物中添加3MHCl溶液(在滤出钯/碳催化剂之后)并搅拌15-20分钟来提纯。所得混合物随后用二乙醚萃取以便从混合物中除去环己酮,留下环己基吡咯烷鎓盐酸盐。将含有环己基吡咯烷鎓盐酸盐的水层分离并随后用NaOH溶液中和至9-10的pH。游离的环己基吡咯烷用乙酸乙酯萃取,在MgSO4上干燥并在旋转蒸发仪上在减压下浓缩以获得不含任何杂质的所需产物。实施例2合成N-(3-甲基环己基)吡咯烷如下面图式2中所示,使用上文实施例1中所描述的程序,使用3-甲基环己酮取代环己酮合成N-(3-甲基环己基)吡咯烷。缩合反应产生了N-(3-甲基环己-1-烯基)吡咯烷和N-(5-甲基环己-1-烯基)吡咯烷的异构体混合物,其在氢化时以88%的产率生成所需N-(3-甲基环己基)吡咯烷。图式2实施例3合成N-甲基-N-环己基吡咯烷鎓氢氧化物参照下面的图式3,将实施例1中制备的N-环己基吡咯烷(1摩尔当量)在配备有机械搅拌器和回流冷凝器的三颈反应烧瓶中在甲醇中溶解至0.5M浓度。向环己基吡咯烷的甲醇溶液中加入2摩尔当量的碘甲烷,所得混合物搅拌整夜。将混合物加热至回流并在回流下搅拌4小时。将反应冷却并搅拌整夜。反应完成。该反应混合物在旋转蒸发仪上浓缩。所得棕褐色固体溶解在异丙醇中,随后通过添加二乙醚从溶液中沉淀出来。将沉淀物过滤出来并在旋转蒸发仪上在减压下和在80℃下的热水浴中干燥,以86%的产率获得所需N-甲基-N-环己基吡咯烷鎓碘化物。通过用氢氧根离子离子交换该碘离子,将N-甲基-N-环己基吡咯烷鎓碘化物转化为氢氧化物。在聚乙烯塑料瓶中,将N-甲基-N-环己基吡咯烷鎓碘化物盐溶解在去离子水(1毫摩尔盐/10毫升H2O)中。随后加入Bio-RadAG1-X8树脂(1.1克树脂/毫摩尔盐),并温和搅拌淤浆状混合物整夜。交换溶液随后过滤,并将小的等分试样用0.1N的HCl滴定以获得N-甲基-N-环己基吡咯烷鎓氢氧化物的92%产率。图式3实施例4合成N-甲基-N-(3-甲基环己基)吡咯烷鎓氢氧化物参照下面的图式4,将上面实施例2中制备的N-(3-甲基环己基)吡咯烷用碘甲烷以类似于实施例3中所述程序的方式季铵化。该季铵化反应以94%的产率提供所需的N-甲基-N-(3-甲基环己基)吡咯烷鎓碘化物。所得碘盐如实施例3中所述用Bio-RadAG1-X8离子交换树脂交换,以98%的产率(滴定分析)获得N-甲基-N-(3-甲基环己基)吡咯烷鎓氢氧化物。图式4实施例5合成N-乙基-N-(3-甲基环己基)吡咯烷鎓氢氧化物如图式5中所示,除了用碘乙烷取代碘甲烷,以类似于实施例5中所述程序的方式用碘乙烷将上面实施例2中制备的N-(3-甲基环己基)吡咯烷季铵化。季铵化以85%的产率获得所需N-乙基-N-(3-甲基环己基)吡咯烷鎓碘化物。所得N-乙基-N-(3-甲基环己基)吡咯烷鎓碘盐如实施例3中所述用Bio-RadAG1-X8离子交换树脂交换,以88%的产率(通过滴定)获得N-甲基-N-(3-甲基环己基)吡咯烷鎓氢氧化物。图式5实施例6使用N-环己基-N-甲基吡咯烷鎓阳离子合成Al-SSZ-13(Al-CHA)在23立方厘米的Teflon内衬中,将3.6克0.63M的N-环己基-N-甲基吡咯烷鎓氢氧化物溶液(2毫摩尔SDA)、3克1NKOH溶液和5.3克Nalco胶态铝硅酸盐(含有~19重量%的固体,SiO2/Al2O3比为~35)混合并用Teflon桨叶搅拌直到获得均匀的凝胶。所得凝胶加盖并密封在高压釜中,所述高压釜固定到烘箱中的旋转器(rotatingspit)(~43rpm)上并在170℃下加热。通过扫描电子显微镜分析和通过监控反应凝胶的pH来追踪结晶进程。在加热7天后反应完成,获得澄清溶液和pH为12.1的微细粉末沉淀物。用烧结玻璃漏斗过滤反应溶液。获得的固体用去离子水(1升)彻底冲洗并空气干燥整夜。随后,固体在烘箱中在125℃下进一步干燥1小时。反应生成0.92克通过SEM和粉末XRD分析所显示的纯SSZ-13(CHA),晶体尺寸为0.5微米。制得后原样的产品的所得XRD图谱显示在图1中。制得后原样的样品在空气中在马弗炉中煅烧,以1℃/分钟的速率由室温加热至120℃,并在120℃下保持2小时。该温度随后以1℃/分钟的速率升至540℃。样品在540℃下保持5小时。温度以相同速率(1℃/分钟)提高至595℃并保持5小时。在煅烧时,存在20%的重量损失。煅烧产物的所得XRD图谱显示在图2中。实施例7重复上面的实施例6,但是这次该反应用0.06克Al-SSZ-13(Al-CHA)作为晶种。反应在5天内完成,获得0.96克SSZ-13。实施例8使用N-甲基-N-(3-甲基环己基)吡咯烷鎓阳离子合成Al-SSZ-13(Al-CHA)除了用3.3克0.69M的N-甲基-N-(3-甲基环己基)吡咯烷鎓氢氧化物溶液替代该SDA之外,完全重复实施例6。该反应在6天内完成,获得0.84克0.5-3微米的纯结晶Al-SSZ-13。该Al-SSZ-13产物通过粉末XRD光谱法分析。制得后原样的产物的所得XRD图谱显示在图3中。制得后原样的产物在在空气中在马弗炉中煅烧,以1℃/分钟的速率由室温加热至120℃,并在120℃下保持2小时。该温度随后以1℃/分钟的速率升至540℃。样品在540℃下保持5小时。温度以相同速率(1℃/分钟)提高至595℃并保持5小时。在煅烧时,存在19%的重量损失。煅烧产物的所得XRD图谱显示在图4中。实施例9重复实施例8,用0.05克Al-SSZ-13作为晶种。反应在4天内完成,获得1克纯CHA(Al-SSZ-13)。实施例10使用N-乙基-N-(3-甲基环己基)吡咯烷鎓阳离子合成Al-SSZ-13(Al-CHA)使用3.5克N-乙基-N-(3-甲基环己基)吡咯烷鎓氢氧化物的0.68M溶液作为结构导向剂重复上面的实施例6。加热后反应在14天完成,获得0.89克纯Al-CHA(Al-SSZ-13),晶体尺寸为0.5-1微米。该Al-SSZ-13产物通过粉末XRD光谱法分析。制得后原样的产物的所得XRD图谱显示在图5中。制得后原样的产物在在空气中在马弗炉中煅烧,以1℃/分钟的速率由室温加热至120℃,并在120℃下保持2小时。该温度随后以1℃/分钟的速率升至540℃。样品在540℃下保持5小时。温度以相同速率(1℃/分钟)提高至595℃并保持5小时。在煅烧时,存在21%的重量损失。煅烧产物的所得XRD图谱显示在图6中。实施例11重复实施例10,用0.07克Al-SSZ-13作为晶种。反应在8天内完成,获得0.98克纯SSZ-13。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1