重组酵母和使用该重组酵母的物质生产方法与流程
本发明涉及以抑制和/或强化的方式改变规定基因的表达,还涉及导入了规定基因的重组酵母、以及使用该重组酵母的物质生产方法。
背景技术:
作为与使用酵母的物质生产相关的技术,主要可列举设计以乙酰辅酶A为中间体的物质生产途径的方法。例如,作为代表性脂肪酸的油酸,需要以9分子的乙酰辅酶A为原料,作为代表性二萜的胡萝卜素,需要以12分子的乙酰辅酶A为原料。这样,已知利用酵母内所蓄积的乙酰辅酶A来合成作为药品和/或精细化学品有用的脂肪酸的技术(专利文献1)、合成萜类的技术(专利文献2)、合成聚酮类化合物的技术(专利文献3)。另外,作为以乙酰辅酶A为中间体合成的物质,可列举作为生物燃料收到关注的丁醇(专利文献4)、异丙醇(专利文献5)和法呢烷(专利文献5)。但是,在酵母中,乙酰辅酶A是在菌体外生产的乙醇被摄入体内,由所摄入的乙醇合成的。如果由酵母生产的乙醇变为高浓度,则酵母自身的生长被抑制。因此,通过提高酵母的乙醇生产能力,或者提高酵母的乙醇摄入量这样的方法来提高菌体内的乙酰辅酶A量是困难的。更具体地,专利文献2公开了由乙酰辅酶A合成法呢烷的技术,但其收率为理论收率的25%左右。另外,专利文献6公开了由乙酰辅酶A合成6-甲基水杨酸的技术,但其收率为理论收率的20%左右。这样,在由乙酰辅酶A的物质生产中,存在生产性显著低这样的问题。现有专利文献专利文献1:日本特开昭63-287491号公报专利文献2:WO2008/045555专利文献3:日本特开2008-22865专利文献4:WO2008/137406专利文献5:US2008/0293125专利文献6:US2006/0148052
技术实现要素:
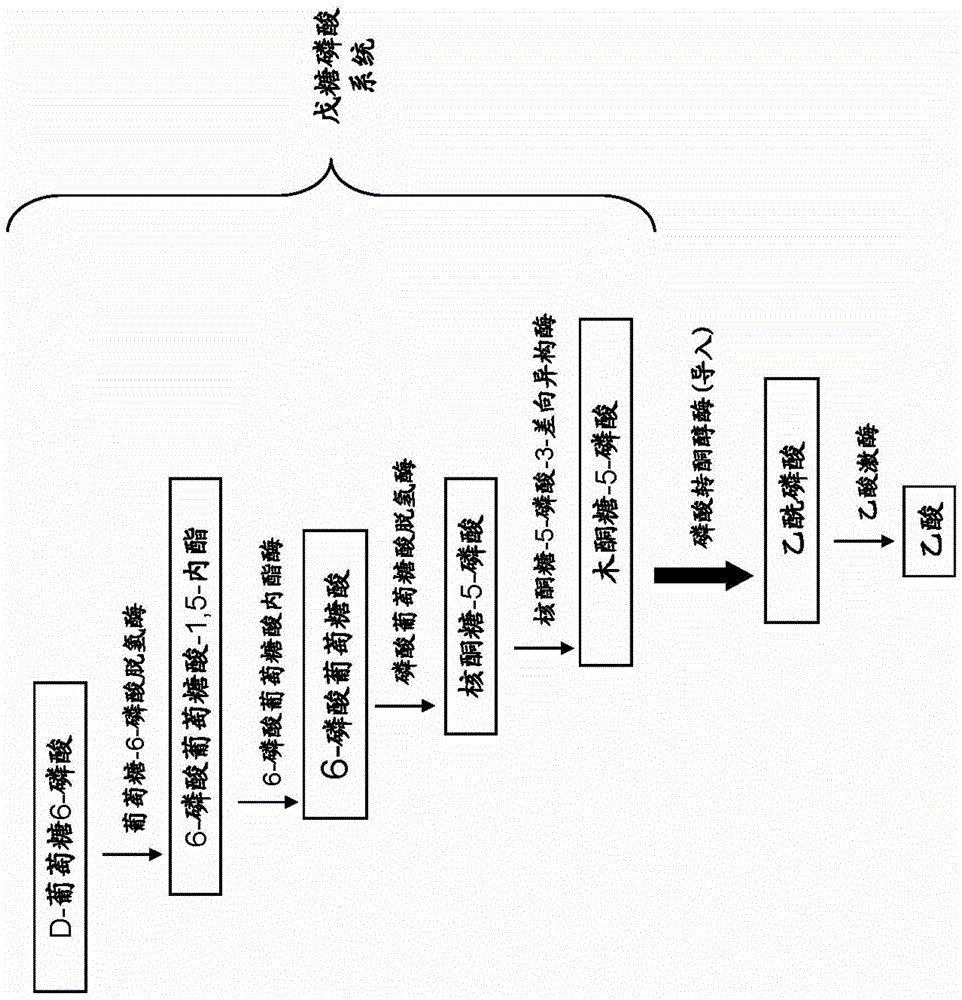
发明要解决的课题于是,本发明鉴于上述事实,目的在于特别通过在酵母中导入由葡萄糖6磷酸合成乙酰辅酶A或乙酸的代谢途径,从而提供物质生产性优异的重组酵母和使用该酵母的物质生产方法。用于解决课题的方法为了实现上述目的,本发明者们进行了深入研究,结果发现,通过对酵母在削弱参与糖酵解系统的基因的同时导入磷酸转酮醇酶基因,从而乙酸、乙酰辅酶A和来自乙酰辅酶A的物质的生产性提高,从而完成了本发明。此外,磷酸转酮醇酶是催化将木酮糖5-磷酸转换成乙酰磷酸和甘油醛3-磷酸的反应的酶。即,本发明包含以下(1)~(7)。(1)重组酵母,其中,削弱了磷酸果糖激酶基因,且导入了磷酸转酮醇酶基因。(2)根据(1)所述的重组酵母,其特征在于,强化了葡萄糖-6-磷酸脱氢酶基因和/或D-核酮糖-5-磷酸-3-差向异构酶基因的表达。(3)根据(1)所述的重组酵母,其特征在于,导入了磷酸转乙酰酶基因,和/或强化了乙酰辅酶A合成酶基因的表达。(4)根据(1)所述的重组酵母,其特征在于,强化了参与以乙酰辅酶A为底物合成乙酸乙酯的反应的醇乙酰转移酶基因的表达。(5)根据(1)所述的重组酵母,其特征在于,导入了参与以乙酰辅酶A为底物合成异丙醇的反应的乙酰乙酸脱羧酶基因、丁酸-乙酰乙酸辅酶A转移酶的亚基A基因、丁酸-乙酰乙酸辅酶A转移酶的亚基B基因、乙酰辅酶A乙酰转移酶基因和异丙醇脱氢酶基因。(6)物质的制造方法,包括将上述(1)~(5)的任一项所述的重组酵母用培养基进行培养的工序。(7)根据(6)所述的物质的制造方法,其特征在于,上述物质是选自由乙酸、乙酰辅酶A、来自乙酰辅酶A的乙酸乙酯和来自乙酰辅酶A的异丙醇组成的组中的1种。发明的效果本发明的重组酵母,将果糖-6-磷酸转换为果糖-1,6-二磷酸的活性被削弱,且被赋予了将木酮糖5-磷酸转换为乙酰磷酸的活性。由此,通过利用本发明的重组酵母,可以提高乙酸和/或来自乙酰辅酶A的物质的生产性。附图说明图1是显示包含在本发明的酵母中削弱的磷酸果糖激酶基因所参与的代谢途径的糖酵解系统的一部分的特性图。图2是显示包含在本发明的酵母中导入的磷酸转酮醇酶基因所参与的代谢途径的戊糖磷酸系统的一部分的特性图。图3是对于来自各种生物的磷酸转酮醇酶基因显示分子系统树分析的结果的特性图。图4是显示由乙酰磷酸和乙酸合成乙酰辅酶A的途径和由乙酰辅酶A合成其他物质的途径的特性图。图5是对于来自各种生物的磷酸转乙酰酶基因显示分子系统树分析的结果的特性图。图6是用于制作pESC-HIS-ZWF1-RPE1载体的流程图。图7是用于制作pESC-Leu-PKT载体和pESC-Leu-PKT-PTA载体的流程图。图8是用于制作pESC-Trp-ATF1载体的流程图。图9是用于制作PFK1基因破坏用载体的流程图。图10是用于制作PFK2基因破坏用载体的流程图。图11是显示乙酸生产试验的结果的特性图。图12是显示乙酸乙酯生产试验的结果的特性图。图13是显示异丙醇生产试验的结果的特性图。图14是显示利用CE-TOFMS的酵母的代谢组分析的结果的特性图。具体实施方式以下使用附图和实施例更详细地说明本发明。本发明的重组酵母,通过削弱编码参与糖酵解系统的酶的基因,并且导入磷酸转酮醇酶基因,而具有将木酮糖5-磷酸转换为乙酰磷酸的活性。作为可以用作宿主的酵母,不特别限定,可列举休哈塔假丝酵母(CandidaShehatae)等假丝酵母(Candida)属酵母、树干毕赤酵母(Pichiastipitis)等毕赤酵母(Pichia)属酵母、嗜鞣管囊酵母(Pachysolentannophilus)等管囊酵母(Pachysolen)属酵母、酿酒酵母(Saccharomycescerevisiae)等酵母(Saccharomyces)属酵母和粟酒裂殖酵母(Schizosaccharomycespombe)等裂殖酵母(Schizosaccharomyces)属酵母,特别优选酿酒酵母。另外,作为酵母,既可以是为了实验方面的方便性而使用的实验株,也可以是为了实用方面的有用性而使用的工业株(实用株)。作为工业株,可列举例如,用于制作红酒、清酒或烧酎的酵母株。这里,作为编码参与糖酵解系统的酶的基因、即削弱对象的基因,可列举磷酸果糖激酶基因。此外,作为参与糖酵解系统的酶,除了磷酸果糖激酶以外,还已知己糖激酶、葡萄糖磷酸异构酶、醛缩酶、丙糖磷酸异构酶、甘油醛-3-磷酸脱氢酶、磷酸甘油酸激酶、磷酸甘油酸变位酶、烯醇化酶和丙酮酸激酶。也可以削弱这些编码磷酸果糖激酶以外的酶的基因。磷酸果糖激酶基因如图1所示,在糖酵解系统中,编码将果糖-6-磷酸转换成果糖-1,6-二磷酸的酶。削弱磷酸果糖激酶基因是指使磷酸果糖激酶活性有意义地降低,换言之,是指使糖酵解系统中的果糖-1,6-二磷酸的合成量有意义地降低。作为削弱磷酸果糖激酶基因的方法,不特别限定,可列举破坏磷酸果糖激酶基因或使其缺失、破坏磷酸果糖激酶基因的表达控制区或使其缺失、添加磷酸果糖激酶的抑制物质(例如柠檬酸)、或者利用siRNA等的RNA干涉的方法,和/或通过反义法来抑制磷酸果糖激酶基因的表达这样的方法。此外,作为酿酒酵母的内在的磷酸果糖激酶基因,已知PFK1基因和PFK2基因(THEJOURNALOFBIOLOGICALCHEMISTRY,Vol.275,No.52,IssueofDecember29,pp.40952-40960,2000)。在作为本发明的重组酵母的宿主利用酿酒酵母的情况下,可以削弱上述PFK1基因和PFK2基因的任一者,也可以削弱两者。此外,对于除酿酒酵母以外的酵母,内在的磷酸果糖激酶基因是公知的,可以参照现有的Genbank、DDBJ、EMBL等数据库来特定。这样,通过特定各种酵母中内在的磷酸果糖激酶基因,可以削弱通过上述的方法和/或手段特定的磷酸果糖激酶基因。另外,本发明的重组酵母通过外源地导入磷酸转酮醇酶基因(PKT基因),从而获得将木酮糖5-磷酸转换成乙酰磷酸的能力。此外,木酮糖5-磷酸是作为酵母本来具有的戊糖磷酸系统中的代谢物质由核酮糖-5-磷酸合成的(图2)。通过磷酸转酮醇酶合成的乙酰磷酸,通过酵母本来具有的乙酸激酶而转换成乙酸。因此,在削弱磷酸果糖激酶基因的同时导入了磷酸转酮醇酶基因的重组酵母中,向培养基的乙酸的分泌生产性大幅提高。作为磷酸转酮醇酶基因,不特别限定,例如,优选使用来自具有异质乳酸发酵的代谢途径的乳酸菌和/或双歧杆菌的磷酸转酮醇酶基因。这里,异质乳酸发酵是指由葡萄糖经过糖酵解系统而生成的丙酮酸不仅被代谢为乳酸,还被代谢为乙醇和/或乙酸和二氧化碳的发酵。在异质乳酸发酵中,由通过磷酸转酮醇酶生成的乙酰磷酸,合成乙醇和/或乙酸。更具体地,作为磷酸转酮醇酶基因,如图3所示,可以使用来自各种微生物的磷酸转酮醇酶基因。此外,分配在图3所示的分子系统树中的编号与微生物的对应关系示于下述表1。表1另外,作为磷酸转酮醇酶基因,优选使用在图3所示的分子系统树中被分类于虚线框内的磷酸转酮醇酶基因。分类于这一组的磷酸转酮醇酶基因主要来自具有异质乳酸发酵的代谢途径的乳酸菌和/或双歧杆菌。即,作为磷酸转酮醇酶基因,如图3所示的分子系统树中被分类于虚线框内那样,优选使用来自作为双歧杆菌的双歧杆菌(Bifidobacterium)属微生物、作为乳酸菌的乳酸菌(Lactobacillus)属微生物和/或明串珠菌(Leuconostoc)属微生物的基因。更详细地,作为在图3所示的分子系统树中被分类于虚线框内的磷酸转酮醇酶基因,可以使用编码包含序列号1~19的任一项所记载的氨基酸序列的磷酸转酮醇酶的基因。序列号1的氨基酸序列是由来自动物双歧杆菌(Bifidobacteriumanimalis)的磷酸转酮醇酶基因所编码的氨基酸序列。序列号2~19所示的氨基酸序列分别依次是由来自长双岐杆菌(Bifidobacteriumlongum)、青春双岐杆菌(Bifidobacteriumadolescentis)、鸡源双歧杆菌(Bifidobacteriumpullorum)、植物乳杆菌(Lactobacillusplantarum)、植物乳杆菌(Lactobacillusplantarum)、戊糖乳杆菌(Lactobacilluspentosus)、清酒乳杆菌(Lactobacillussakei)、唾液乳杆菌(Lactobacillussalivarius)、罗伊氏乳杆菌(Lactobacillusreuteri)、约氏乳杆菌(Lactobacillusjohnsonii)、干酪乳杆菌(Lactobacilluscasei)、凝结芽孢杆菌(Bacilluscoagulans)、肠膜明串珠菌(Leuconostocmesenteroides)、肠膜明串珠菌(Leuconostocmesenteroides)、格氏链球菌(Streptococcusgordonii)、丙酮丁醇梭菌(Clostridiumacetobutylicum)、丁酸梭菌(Clostridiumbutyricum)和无乳支原体(mycoplasmaagalactiae)的磷酸转酮醇酶基因所编码的氨基酸序列。即,在本发明中,优选使用编码序列号1~19的任一项所记载的氨基酸序列的磷酸转酮醇酶基因。其中,作为磷酸转酮醇酶基因,最优选使用动物双歧杆菌(Bifidobacteriumanimalis)(序列号1)、长双岐杆菌(Bifidobacteriumlongum)(序列号2)、青春双岐杆菌(Bifidobacteriumadolescentis)(序列号3)和鸡源双歧杆菌(Bifidobacteriumpullorum)(序列号4)。此外,作为磷酸转酮醇酶基因,也可以是包含编码下述蛋白质的多核苷酸的基因,所述蛋白质包含在序列号1~19的任一氨基酸序列中缺失、替换、添加或插入了1或数个氨基酸的氨基酸序列,且具有磷酸转酮醇酶活性。这里,数个氨基酸是指例如2~100个、优选为2~80个、更优选为2~55个、进一步优选为2~15个氨基酸。另外,作为磷酸转酮醇酶基因,还可以是包含编码下述蛋白质的多核苷酸的基因,所述蛋白质包含相对于序列号1~19的任一氨基酸序列具有80%以上、优选为85%以上、更优选为90%以上、进一步优选为98%以上的序列类似性的氨基酸序列,且具有磷酸转酮醇酶活性。这里,序列类似性是指以默认的设定使用BLAST、PSI-BLAST、HMMER等序列类似性检索软件作为显示2个氨基酸序列之间的类似性的值而算出的值。这里,磷酸转酮醇酶活性是将木酮糖-5-磷酸转换为乙酰磷酸的活性。因此,规定的蛋白质是否具有磷酸转酮醇酶活性,可以利用含有木酮糖-5-磷酸作为底物的反应液通过乙酰磷酸的合成量来判定(例如,JOURNALOFBACTERIOLOGY,Vol.183,No.9,May2001,p.29292936)。另外,本发明的重组酵母可以通过强化参与图2所示的戊糖磷酸系统的酶基因的表达,来提高利用导入的磷酸转酮醇酶基因的乙酰磷酸的合成量,其结果可以提高乙酸的合成量。作为在图2所示的戊糖磷酸系统中强化表达的对象的基因,不特别限定,可列举葡萄糖-6-磷酸脱氢酶基因和核酮糖-5-磷酸-3-差向异构酶基因。此外,既可以强化这些基因中的一种基因的表达,也可以强化两种基因的表达。此外,作为酿酒酵母中内在的葡萄糖-6-磷酸脱氢酶基因,已知ZWF1基因。另外,作为酿酒酵母中内在的核酮糖-5-磷酸-3-差向异构酶基因,已知RPE1基因。对于除酿酒酵母以外的酵母,内在的葡萄糖-6-磷酸脱氢酶基因和/或核酮糖-5-磷酸-3-差向异构酶基因也是公知的,可以参照现有的Genbank、DDBJ、EMBL等数据库来特定。这里,强化基因的表达是指有意义地提高由成为对象的基因所编码的酶的活性,是包含有意义地提高该基因的表达量的含义。作为强化基因的表达的方法,可以列举有意义地提高该基因的表达量的方法。作为提高特定的基因的表达量的方法,不特别限定,可列举改变染色体中内在的基因的表达控制区的方法、导入在活性高的启动子的下游配置了该基因的载体的方法。另一方面,本发明的重组酵母除了磷酸果糖激酶基因的削弱和磷酸转酮醇酶基因的导入之外,如图4所示,可以通过进一步导入磷酸转乙酰酶基因(PTA基因),或进一步强化乙酰辅酶A合成酶基因(ACS基因),来提高乙酰辅酶A的合成量。磷酸转乙酰酶基因是本来酵母不具有的基因,作为外来基因而被导入。作为磷酸转乙酰酶基因,不特别限定,可以广泛应用在各种细菌中被称为PTA基因的基因。更具体地,作为磷酸转乙酰酶基因,如图5所示,可以使用来自各种微生物的磷酸转乙酰酶基因。此外,分配在图5所示的分子系统树中的编号与微生物的对应关系示于下述表2。表21Bacillus_subtilis2Bacillus_amyloliquefaciens3Bacillus_licheniformis4Geobacillus_thermodenitrificans5Listeria_innocua6Staphylococcus_aureus7Lactococcus_lactis8Carnobacterium_sp.9Mycobacterium_vanbaalenii10Clostridium_perfringens11Enterococcus_faecalis12Leuconostoc_mesenteroides13Clostridium_acetobutylicum14Bifidobacterium_animalis_lactis15Corynebacterium_glutamicum16Escherichia_coli_K-1217Escherichia_coli_5363818Vibrio_vulnificus19Haemophilussomnus20Yersinia_pestis21Shigella_sonnei将由来自图5和表2所示的枯草芽孢杆菌(Bacillussubtilis)的PTA基因所编码的蛋白质的氨基酸序列示于序列号20,将由来自解淀粉芽孢杆菌(Bacillusamyloliquefaciens)的PTA基因所编码的蛋白质的氨基酸序列示于序列号21,将由来自地衣芽孢杆菌(Bacilluslicheniformis)的PTA基因所编码的蛋白质的氨基酸序列示于序列号22,由来自嗜热脱氮土壤芽孢杆菌(Geobacillusthermodenitrificans)的PTA基因所编码的蛋白质的氨基酸序列示于序列号23,由来自无害李斯特菌(Listeriainnocua)的PTA基因所编码的蛋白质的氨基酸序列示于序列号24,由来自金黄色葡萄球菌(Staphylococcusaureus)的PTA基因所编码的蛋白质的氨基酸序列示于序列号25,由来自乳酸乳球菌(Lactococcuslactis)的PTA基因所编码的蛋白质的氨基酸序列示于序列号26,由来自肉杆菌属(Carnobacteriumsp.)的PTA基因所编码的蛋白质的氨基酸序列示于序列号27,由来自Mycobacteriumvanbaalenii的PTA基因所编码的蛋白质的氨基酸序列示于序列号28,由来自产气荚膜梭菌(Clostridiumperfringens)的PTA基因所编码的蛋白质的氨基酸序列示于序列号29,由来自粪肠球菌(Enterococcusfaecalis)的PTA基因所编码的蛋白质的氨基酸序列示于序列号30,由来自肠膜明串珠菌(Leuconostocmesenteroides)的PTA基因所编码的蛋白质的氨基酸序列示于序列号31,由来自丙酮丁醇梭菌(Clostridiumacetobutylicum)的PTA基因所编码的蛋白质的氨基酸序列示于序列号32,由来自动物双歧杆菌乳亚种(Bifidobacteriumanimalis_lactis)的PTA基因所编码的蛋白质的氨基酸序列示于序列号33,由来自谷氨酸棒杆菌(Corynebacteriumglutamicum)的PTA基因所编码的蛋白质的氨基酸序列示于序列号34,由来自大肠杆菌(Escherichiacoli)K-12的PTA基因所编码的蛋白质的氨基酸序列示于序列号35,由来自大肠杆菌(Escherichiacoli)53638的PTA基因所编码的蛋白质的氨基酸序列示于序列号36,由来自创伤弧菌(Vibriovulnificus)的PTA基因所编码的蛋白质的氨基酸序列示于序列号37,由来自睡眠嗜血杆菌(Haemophilussomnus)的PTA基因所编码的蛋白质的氨基酸序列示于序列号38,由来自鼠疫耶尔森氏菌(Yersiniapestis)的PTA基因所编码的蛋白质的氨基酸序列示于序列号39,由来自索氏志贺菌(Shigellasonnei)的PTA基因所编码的蛋白质的氨基酸序列示于序列号40。此外,作为磷酸转乙酰酶基因,也可以是包含编码下述蛋白质的多核苷酸的基因,所述蛋白质包含在序列号20~40的任一氨基酸序列中缺失、替换、添加或插入了1或数个氨基酸的氨基酸序列,且具有磷酸转乙酰酶活性。这里,数个氨基酸是指例如2~35个、优选为2~25个、更优选为2~15个、进一步优选为2~10个氨基酸。另外,作为磷酸转乙酰酶基因,也可以是包含编码下述蛋白质的多核苷酸的基因,所述蛋白质包含相对于序列号20~40的任一氨基酸序列具有80%以上、优选为85%以上、更优选为90%以上、进一步优选为98%以上的序列类似性的氨基酸序列,且具有磷酸转乙酰酶活性。这里,序列类似性是指以默认的设定使用BLAST、PSI-BLAST、HMMER等序列类似性检索软件作为显示2个氨基酸序列之间类似性的值而算出的值。这里,磷酸转乙酰酶活性是指向乙酰磷酸转移辅酶A的活性。因此,规定的蛋白质是否具有磷酸转乙酰酶活性,可以利用含有乙酰磷酸和辅酶A的反应液通过乙酰辅酶A的合成量来判定。另外,图4所示的乙酰辅酶A合成酶基因是酵母本来具有的基因。因而为了强化乙酰辅酶A合成酶基因,可以应用改变染色体内在的该基因的表达控制区的方法、导入在活性高的启动子的下游配置了该基因的载体的方法。此外,作为酿酒酵母中内在的乙酰辅酶A合成酶基因,已知ACS1基因和ACS2基因。对于除酿酒酵母以外的酵母,内在的乙酰辅酶A合成酶基因也是公知的,可以参照现有的Genbank、DDBJ、EMBL等数据库来特定。如上,本发明的重组酵母,乙酰磷酸的合成量即乙酸的合成量显著提高(图2),或者乙酰辅酶A的合成量显著提高(图4)。因此,本发明的重组酵母可以在以乙酸或乙酰辅酶A为生产对象时使用。或者,本发明的重组微生物可以为了能够以乙酰辅酶A为底物制造其他物质(图4中表述为来自乙酰辅酶A的物质)而作为用于进行进一步改变的宿主使用。具体地,作为来自乙酰辅酶A的物质,不特别限定,可以合成丁醇、烷烃、丙醇、脂肪酸、脂肪酸酯、丙酮、乙酰乙酸、乙酸乙酯、聚酮化合物、氨基酸和萜类。以这些作为生产对象的物质时,通过利用本发明的重组酵母,可以大幅提高生产对象的物质的生产性。以异丙醇作为生产对象物质时,例如,可以通过参照APPLIEDANDENVIRONMENTALMICROBIOLOGY,Dec.2007,p.7814-7818,Vol.73,No.24来特定应在本发明的重组微生物中进一步导入的基因。另外,以聚酮类化合物作为生产对象物质时,可以通过参照Proc.Natl.Acad.Sci.USA,Vol.95,pp.505-509,January1998来特定应在本发明的重组微生物中进一步导入的基因。进而,在以脂肪酸为生产对象物质时,例如,可以通过参照Eur.J.Biochem.112,p.431-442(1980)或MICROBIOLOGYANDMOLECULARBIOLOGYREVIEWS,Sept.2004,p.501-517来特定应在本发明的重组微生物中进一步导入、或强化的基因(例如,FAS基因)。进一步,在以烷烃作为生产对象物质时,对于参与由脂肪酸合成醛、进一步由醛合成烷烃的基因,例如,可以通过参照Sciencevol32930julyp559-562来特定应在本发明的重组微生物中进一步导入的基因。另外,通过进一步强化酵母中内在的醇乙酰转移酶基因(ATF1基因)的表达,可以提高由乙酰辅酶A的乙酸乙酯的合成量。即,以乙酸乙酯作为生产对象物质时,优选强化内在的ATF1基因的表达。另外,如上所述,在强化规定基因的表达时,使用转录活性高的适当启动子。作为这样的启动子,不特别限定,可以利用例如甘油醛3磷酸脱氢酶基因(TDH3)的启动子、3-磷酸甘油酸酯激酶基因(PGK1)的启动子、高渗透压应答7基因(HOR7)的启动子等。其中丙酮酸脱羧酶基因(PDC1)的启动子使下游的目的基因高表达的能力高,因此优选。此外,通过使用gal1启动子、gal10启动子、热休克蛋白质启动子、MFα1启动子、PHO5启动子、GAP启动子、ADH启动子、AOX1启动子等,可以使下游的基因强表达。另外,作为导入上述的基因的方法,还可以应用作为酵母的转化方法已知的现有公知的任何方法。具体地,例如,可以通过电穿孔法“Meth.Enzym.,194,p182(1990)”、原生质球法“Proc.Natl.Acad.Sci.USA,75p1929(1978)”、乙酸锂法“J.Bacteriology,153,p163(1983)”、Proc.Natl.Acad.Sci.USA,75p1929(1978)、Methodsinyeastgenetics,2000Edition:AColdSpringHarborLaboratoryCourseManual等所记载的方法来实施,但不限于此。在使用以上所说明的重组酵母来制造物质时,用含有适当的碳源的培养基进行培养。更具体地,按照常规方法用培养基培养进行了前培养的重组酵母,生产目的物质。例如,制造丁醇、烷烃、丙醇、脂肪酸、脂肪酸酯、丙酮、乙酰乙酸、乙酸酯、聚酮化合物、氨基酸和萜类作为目的物质时,这些目的物质在培养基中合成,可以通过离心分离等手段,由从培养基分离菌体后的上清级分中分离物质。为了由上清级分中分离物质,例如,可以在上清级分中添加乙酸乙酯和甲醇等有机溶剂,充分搅拌。分离成水层和溶剂层,可以从溶剂层提取物质。实施例以下,通过实施例更详细地说明本发明,但本发明的技术范围不限于以下实施例。在本实施例中,制作在野生型的酵母和异丙醇生产酵母中在削弱内在的磷酸果糖激酶基因的同时导入磷酸转酮醇酶基因而得的重组酵母、和进一步导入或强化了其他基因的重组酵母,对于乙酸、乙酸乙酯和异丙醇的生产性进行研究。〔材料和方法〕宿主使用ECOSCompetentE.coliJM109(ニッポンジーン社制)、作为野生型酵母的酿酒酵母(S.cerevisiae)YPH499(stratagene社制)和作为异丙醇生产酵母的后述参考例所公开的#15-10株。质粒<pESCpgkgap-HIS的制作>在以下条件下进行PCR。(引物)EcoRI-Pgap-F:5’-CACGGAATTCCAGTTCGAGTTTATCATTATCAA-3’(序列号41)BamHI-Pgap-R:5’-CTCTGGATCCTTTGTTTGTTTATGTGTGTTTATTC-3’(序列号42)(PCR条件)模板:pDI626质粒1ng(参照日本特开2005-52046号公报)引物:50pmol引物DNA,反应液:包含1xPfuUltraIIreactionbuffer(Stratagene社制)、10nmoldNTP和1μlPfuUltraIIfusionHSDNApolymerase(Stratagene社制)的50μl溶液反应:95℃2分钟-(95℃30秒、55℃30秒、72℃2分钟)×25个循环-72℃3分钟-4℃存放在上述条件下进行PCR后,使用QIAGEN社制的MinElutePCRpurificationkit来纯化反应液所含的PCR产物。然后,将PCR产物用限制性酶BamHI和EcoRI消化。进行琼脂糖凝胶电泳,切下686bp的片段并使用QIAGEN社制MiniEluteGelextractionkit进行纯化。另外,与用限制性酶BamHI和EcoRI消化后的pESC-HIS载体连接。将这样得到的质粒命名为pESCgap-HIS。接着在以下条件下进行PCR。(引物)MunI-Ppgk1-F:5’-TAGGCAATTGCAAGAATTACTCGTGAGTAAGG-3’(序列号43)EcoRI-Ppgk1-R:5’-ATAAGAATTCTGTTTTATATTTGTTGTAAAAAGTAG-3’(序列号44)(PCR条件)模板:pDI626PGK质粒1ng引物:50pmol引物DNA反应液:包含1xPfuUltraIIreactionbuffer(Stratagene社制)、10nmoldNTP和1μlPfuUltraIIfusionHSDNApolymerase(Stratagene社制)的50μl溶液反应:95℃2分钟-(95℃30秒、55℃30秒、72℃2分钟)×25个循环-72℃3分钟-4℃存放在上述条件下进行PCR后,使用QIAGEN社制MinElutePCRpurificationkit来纯化反应液所含的PCR产物。然后,将PCR产物用限制性酶MunI和EcoRI消化。进行琼脂糖凝胶电泳,切下718bp的片段并使用QIAGEN社制MinEluteGelextractionkit进行纯化。另外,与经限制性酶EcoRI消化、BAP处理的pESCgap-HIS载体连接。将这样得到的质粒命名为pESCpgkgap-HIS。<pESCpgkgap-LEU的制作>将上述pESCpgkgap-HIS用限制性酶BamHI和NotI消化后,切下1427bp的片段,与同样地用限制性酶BamHI和NotI消化后的pESC-LEU载体(Stratagene社制)连接。<pESCpgkgap-TRP的制作>将上述pESCpgkgap-HIS用限制性酶BamHI和NotI消化后,切下1427bp的片段,与同样地用限制性酶BamHI和NotI消化后的pESC-TRP载体(Stratagene社制)连接。<pESCpgkgap-URA的制作>将上述pESCpgkgap-HIS用限制性酶BamHI和NotI消化后,切下1427bp的片段,与同样地用限制性酶BamHI和NotI消化后的pESC-URA载体(Stratagene社制)连接。<其他载体的制作>在制作用于强化ZWF1基因和RPE1基因的表达的pESC-HIS-ZWF1-RPE1载体、用于导入PKT基因的pESC-Leu-PKT载体、用于导入PKT基因和PTA基因的pESC-Leu-PKT-PTA载体、用于强化ATF1基因的表达的pESC-Trp-ATF1载体、和PFK1基因以及PFK2基因破坏用载体时,在以下的组成和条件下进行PCR。此外,DNA聚合物使用KOD-Plus-Ver.2(TOYOBO社制)。表3<组成>表4<PCR条件>94℃2分钟→98℃10秒、Tm-5℃30秒、68℃5分钟(×30)→68℃5分钟将制作pESC-HIS-ZWF1-RPE1载体的流程示于图6。将制作pESC-Leu-PKT载体和pESC-Leu-PKT-PTA载体的流程示于图7。将制作pESC-Trp-ATF1载体的流程示于图8。将制作PFK1基因破坏用载体的流程示于图9。将制作PFK2基因破坏用载体的流程示于图10。在图6~10所示的载体制作流程中使用的引物归纳于表5。表5此外,PCR产物的纯化使用QIAquickPCRPurificationKit(QIAGEN社制)。另外,在图6~10所示的载体制作流程中,PCR产物的TOPO克隆使用ZeroBluntTOPOPCRcloningkit(invitrogen社制)。另外,pAUR135由タカラバイオ(株)购入。在图6~10所示的载体制作流程中,使用QIAquickGelExtractionKit(QIAGEN社制)从琼脂糖凝胶切下DNA片段。在图6~10所示的载体制作流程中,载体与DNA片段的连接通过利用Ligation-convenienceKit(ニッポンジーン社制)的连接反应、或利用In-FusionDry-DownPCRCloningKit(クロンテック社制)的注入反应来进行。另外,在图7所示的载体制作流程中,表述为pBSK-PKT和pBSK-PTA的载体是合成5种PKT基因和3种PTA基因并插入pBluescriptIISK(+)的SmaI位点而制作的。此外,这5种PKT基因和3种PTA基因全部最优化成酿酒酵母用密码子。5种PKT基因是来自动物双歧杆菌乳亚种(Bifidobacteriumanimalissubsp.lactis)的磷酸转酮醇酶基因(序列号63和64)、来自米曲霉(Aspergillusoryzae)RIB40的磷酸转酮醇酶I基因(序列号65和66)、来自米曲霉(Aspergillusoryzae)RIB40的磷酸转酮醇酶II基因(序列号67和68)、来自点状念珠藻(Nostocpunctiforme)ATCC29133的磷酸转酮醇酶基因(序列号69和70)、来自异养型水油海杆菌(Marinobacteraquaeolei)ATCC700491的磷酸转酮醇酶基因(序列号71和72)。另外,3种PTA基因是来自枯草芽孢杆菌枯草亚种168株(Bacillussubtilissubsp.subtilisstr.168)的磷酸转乙酰酶基因(序列号73和74)、来自动物双歧杆菌乳亚种(Bifidobacteriumanimalissubsp.lactis)AD011的磷酸转乙酰酶基因(序列号75和76)和来自大肠杆菌(Escherichiacoli)K-12MG1655的磷酸转乙酰酶基因(序列号77和78)。转化大肠杆菌的转化按照ECOSCompetentE.coliJM109(ニッポンジーン社制)所附带的方案进行。酵母的转化按照Frozen-EZYeastTransformationIIKit(ザイモリサーチ社制)所附带的方案进行。使用PFK1基因以及PFK2基因破坏用载体的酵母的基因破坏按照pAUR135DNA(タカラバイオ社制)所附带的方案进行。乙酸生产试验转化体的培养如下进行:在SD-His,Leu琼脂培养基中制作活性菌落,在装入了15ml容量试验管中的2ml的SD-His,Leu培养基中接菌并在30℃进行一夜培养(オリエンタル技研工业制IFM型,130转/分钟)得到前培养液,将前培养液以1%容量接种于装入了500ml容量三角烧瓶中的100ml的SD-His,Leu培养基中进行培养。将培养液进行离心(6000×g、15分钟、室温),将其上清1ml加入玻璃制小玻璃瓶中,将其作为分析用样品。乙酸乙酯生产试验转化体的培养如下进行,在SD-His,Leu,Trp琼脂培养基中制作活性菌落,接种于装入了15ml容量试验管中的2ml的SD-His,Leu,Trp培养基并在30℃进行一夜培养(130转/分钟)得到前培养液,将前培养液以1%容量接种于装入了500ml容量三角烧瓶中的100ml的SD-His,Leu,Trp培养基中。将培养液进行离心(6000×g、15分钟、室温),将其上清1ml装入玻璃制小玻璃瓶中,将其作为分析用样品。异丙醇生产试验转化体的培养如下进行:在SD-His,Leu,Ura,Trp琼脂培养基中制作活性菌落,接种于装入了15ml容量试验管中的2ml的SD-His,Leu,Ura,Trp培养基中并在30℃进行一夜培养(130转/分钟)得到前培养液,将前培养液以1%容量接种于装入了500ml容量三角烧瓶中的50ml的SD-His,Leu,Ura,Trp培养基中进行培养。将培养液进行离心(6000×g、15分钟、室温),将其上清1ml装入玻璃制小玻璃瓶中,将其作为分析用样品。GC分析条件标准品使用乙酸(ナカライテスク社制)、乙酸乙酯(ナカライテスク社制)、异丙醇(ナカライテスク社制)。分析仪器、分析条件对于全部乙酸生产试验、乙酸乙酯生产试验和异丙醇生产试验为以下条件。表6利用CE-TOFMS的酵母的代谢组分析<前处理>在SD-His,Leu,Ura,Trp培养基中进行菌体的培养(30℃),加样使得样品量为15OD单位。将其通过过滤迅速抽滤。接着用Milli-Q水10mL抽滤、洗涤2次。将在过滤器上被集菌的酵母菌体浸渍于含有内标物质5μM的甲醇2mL中,然后将1.6mL移至沉降管。在其中加入1600μL的氯仿和640μL的Milli-Q水进行搅拌,进行离心分离(2,300×g、4℃、5分钟)。离心分离后,移取水相至6根250μL超滤管(MILLIPORE社制、ウルトラフリーMCUFC3LCC离心式过滤器元件5KDa)中。将其离心(9,100×g、4℃、120分钟),进行超滤处理。使滤液干固,再溶解于50μL的Milli-Q水中供测定。<测定>本试验中阴离子性代谢物质(阴离子模式)的测定在以下所示的条件(参照参考文献1)~3))下进行。装置:AgilentCE-TOFMSsystem(AgilentTechnologies社)Capillary:Fusedsilicacapillaryi.d.50μm×80cm测定条件:Runbuffer:AnionBufferSolution(p/n:H3302-1021)Rinsebuffer:AnionBufferSolution(p/n:H3302-1022)Sampleinjection:Pressureinjection50mbar,25secCEvoltage:Positive,30kVMSionization:ESINegativeMScapillaryvoltage:3,500VMSscanrange:m/z50-1,000Sheathliquid:HMTSheathLiquid(p/n:H3301-1020)<参考文献、资料>1)T.Soga,D.N.Heiger:Aminoacidanalysisbycapillaryelectrophoresiselectrosprayionizationmassspectrometry.Anal.Chem.72:1236-1241,2000.2)T.Soga,Y.Ueno,H.Naraoka,Y.Ohashi,M.Tomitaetal.:Simultaneousdeterminationofanionicintermediatesfor枯草芽孢杆菌(Bacillussubtilis)metabolicpathwaysbycapillaryelectrophoresiselectrosprayionizationmassspectrometry.Anal.Chem.74:2233-2239,2002.3)T.Soga,Y.Ohashi,Y.Ueno,H.Naraoka,M.Tomitaetal.:Quantitativemetabolomeanalysisusingcapillaryelectrophoresismassspectrometry.J.ProteomeRes.2:488-494,2003.4)http://vanted.ipk-gatersleben.de/〔结果〕乙酸生产试验供乙酸生产试验的酵母归纳于表7,试验结果示于图11。表7表中,PKT(1)是来自动物双歧杆菌乳亚种(Bifidobacteriumanimalissubsp.lactis)的磷酸转酮醇酶基因,PKT(2)是来自米曲霉(Aspergillusoryzae)RIB40的磷酸转酮醇酶I基因,PKT(3)是来自米曲霉(Aspergillusoryzae)RIB40的磷酸转酮醇酶II基因,PKT(4)是来自点状念珠藻(Nostocpunctiforme)ATCC29133的磷酸转酮醇酶基因,PKT(5)是来自异养型水油海杆菌(Marinobacteraquaeolei)ATCC700491的磷酸转酮醇酶基因。由图11所示的结果可明确,通过在削弱成为宿主的酵母中内在的磷酸果糖激酶基因的同时导入磷酸转酮醇酶基因,提高向不是糖酵解系统而是戊糖磷酸系统(图2)的流量,能够高生产乙酸。另外,作为磷酸转酮醇酶基因,来自双歧杆菌的基因、特别是来自动物双歧杆菌(Bifidobacteriumanimalis)的基因在乙酸的生产性方面优异。由该结果可明确,作为磷酸转酮醇酶基因,在图3所示的系统树中位于虚线框内的磷酸转酮醇酶基因在乙酸生产性的方面更优选。进而,由图11所示的结果可明确,在为了削弱向糖酵解系统的流量而削弱磷酸果糖激酶基因的情况下,优选破坏PFK1基因和PFK2基因两者。此外,明确了可以通过强化参与戊糖磷酸系统(图2)的酶基因,从而进一步提高向戊糖磷酸系统(图2)的流量,从而更高生产乙酸。乙酸乙酯生产试验供乙酸乙酯生产试验的酵母归纳于表8,试验结果示于图12。表8表中,PTA(1)是来自枯草芽孢杆菌枯草亚种168株(Bacillussubtilissubsp.subtilisstr.168)的磷酸转乙酰酶基因,PTA(2)是来自动物双歧杆菌乳亚种(Bifidobacteriumanimalissubsp.lactis)AD011的磷酸转乙酰酶基因,PTA(3)是来自大肠杆菌(Escherichiacoli)K-12MG1655的磷酸转乙酰酶基因。此外,在本试验中,PKT基因使用来自动物双歧杆菌乳亚种(Bifidobacteriumanimalissubsp.lactis)的磷酸转酮醇酶基因。如图12所示,在野生株(YPH499株)中导入了ATF1基因的对照株中,与野生株相比乙酸乙酯的生产性也提高。但是,判明了通过在削弱成为宿主的酵母中内在的磷酸果糖激酶基因的同时导入磷酸转酮醇酶基因,进一步导入磷酸转乙酰酶基因(PTA基因)或强化乙酰辅酶A合成酶基因(ACS基因),与对照相比乙酸乙酯的生产性进一步提高。由该结果和图4所示的概略代谢图谱显示,通过在削弱内在的磷酸果糖激酶基因的同时导入磷酸转酮醇酶基因,进一步导入磷酸转乙酰酶基因(PTA基因)或强化乙酰辅酶A合成酶基因(ACS基因),乙酰辅酶A的合成量显著提高。并且显示,合成的乙酰辅酶A通过ATF1酶而作为乙酸乙酯在菌体外高蓄积。特别是,判明了作为导入的磷酸转乙酰酶基因,来自枯草芽孢杆菌(Bacillussubtilis)的基因从乙酰辅酶A生产性的观点出发是优选的。另外,判明了强化的乙酰辅酶A合成酶基因,在ACS1基因和ACS2基因中,更优选ACS1基因。进而,由图12所示的结果可知,在为了减弱向糖酵解系统的流量而削弱磷酸果糖激酶基因的情况下,优选破坏PFK1基因和PFK2基因两者。此外,明确了通过强化参与戊糖磷酸系统(图2)的酶基因,可以进一步提高向戊糖磷酸系统(图2)的流量,从而更高生产乙酸乙酯。异丙醇生产试验供异丙醇生产试验的酵母归纳于表9,试验结果示于图13。表9表中,PTA(1)~(3)表示与表8相同的基因。如图13所示,判明了通过在野生株(YPH499株)中导入ctfA基因、ctfB基因、adc基因和ipdh基因,可以赋予酵母以异丙醇生产能力。并且判明了,通过在削弱内在的磷酸果糖激酶基因的同时导入磷酸转酮醇酶基因,进一步导入磷酸转乙酰酶基因(PTA基因)或强化乙酰辅酶A合成酶基因(ACS基因),与对照相比异丙醇的生产性显著提高。由该结果和图4所示的概略代谢图谱显示,通过在削弱内在的磷酸果糖激酶基因的同时导入磷酸转酮醇酶基因,进一步导入磷酸转乙酰酶基因(PTA基因)或强化乙酰辅酶A合成酶基因(ACS基因),乙酰辅酶A的合成量显著提高。并且显示,合成的乙酰辅酶A通过ctfA、ctfB、adc和ipdh而作为异丙醇在菌体外高蓄积。特别是判明了作为导入的磷酸转乙酰酶基因,来自枯草芽孢杆菌(Bacillussubtilis)的基因从乙酰辅酶A生产性的观点出发是优选的。另外明确了,强化的乙酰辅酶A合成酶基因在ACS1基因和ACS2基因中更优选ACS1基因。利用CE-TOFMS的酵母的代谢组分析对于在削弱野生型(YPH499株)、YPH499株中内在的磷酸果糖激酶基因的同时强化了参与戊糖磷酸系统(图2)的酶基因的株(表述为YPH499ΔPFKα/ZWF-RPE)、和在削弱YPH499株中内在的磷酸果糖激酶基因的同时强化了参与戊糖磷酸系统(图2)的酶基因、并导入了磷酸转酮醇酶基因和磷酸转乙酰酶基因的株(表述为YPH499ΔPFKα/ZWF-RPE,PKT-PTA),将代谢组分析的结果示于图14。图14中,关于YPH499株(野生株)的乙酰辅酶A量,是介由乙醇的乙酰辅酶A合成途径和线粒体内的乙酰辅酶A合成途径的和。此外,酵母中乙酰辅酶A合成途径存在上述介由乙醇的乙酰辅酶A合成途径和线粒体内的乙酰辅酶A合成途径2种途径。另一方面,在YPH499ΔPFKα/ZWF-RPE株中,介由乙醇的乙酰辅酶A合成途径不发挥作用。这是因为,由于ZWF而NADP被消耗,其结果造成催化由醛向乙酸的反应的ALD2不发挥作用(参照乙酸生产试验(图11)的对照株中的乙酸生产性)。因此,YPH499ΔPFKα/ZWF-RPE株中的乙酰辅酶A合成量的值对应于线粒体内的乙酰辅酶A合成途径中生成的乙酰辅酶A量。此外,线粒体内合成的乙酰辅酶A在线粒体内被消耗,因而不能作为乙酸乙酯、异丙醇等物质的原料使用。因此,如果不能在酵母细胞的胞质溶胶中提高乙酰辅酶A的合成量,则不能提高使用合成的乙酰辅酶A的物质的合成量。而且,YPH499ΔPFKα/ZWF-RPE,PKT-PTA株是通过在YPH499ΔPFKα/ZWF-RPE株中导入PKT基因和PTA基因而构建了新的乙酰辅酶A合成途径(参照图2和4)的突变株。因此,通过从YPH499ΔPFKα/ZWF-RPE,PKT-PTA株中的乙酰辅酶A合成量减去YPH499ΔPFKα/ZWF-RPE株中的乙酰辅酶A合成量,可以评价利用上述的新的乙酰辅酶A合成途径的乙酰辅酶A的合成量。详细地,由图14所示的结果而得到了以下结论:通过新的乙酰辅酶A合成途径,可以合成相当于62-14=48pmol的乙酰辅酶A。〔参考例〕在本参考例中,对于上述的实施例中使用的异丙醇生产酵母(#15-10)的制作进行说明。异丙醇合成基因的取得将以下的4个基因由作为梭菌的丙酮丁醇梭菌(Clostridiumacetobutylicum)ATCC824株的基因组克隆到pT7Blue载体中。adc(乙酰乙酸脱羧酶,Acetoacetatedecarboxylase)ctfA(丁酸-乙酰乙酸辅酶A-转移酶亚基A,Butyrate-acetoacetateCoA-transferasesubunitA)ctfB(丁酸-乙酰乙酸辅酶A-转移酶亚基B,Butyrate-acetoacetateCoA-transferasesubunitB)thiA(乙酰辅酶A乙酰转移酶,Acetyl-CoAacetyltransferase)pT7Blue-ADC的制作在以下的条件下进行PCR。引物adc-F:5’-ATGTTAAAGGATGAAGTAATTAAACAAATTAG-3’(序列号79)adc-R:5’-TTACTTAAGATAATCATATATAACTTCAGCTC-3’(序列号80)反应条件模板:0.4μg的梭菌的基因组DNA引物:50pmol引物DNA,反应液:包含1xPfuUltraIIreactionbuffer(Stratagene社制)、10nmoldNTP、1μlPfuUltraIIfusionHSDNApolymerase(Stratagene社制)的50μl溶液反应:95℃5分钟-(95℃30秒、60℃30秒、72℃2分钟)×30个循环-72℃3分钟-4℃存放将扩增的735bp的片段使用Novagen社制PerfectlyBluntCloningKit平端克隆pT7Blue载体中。对克隆的序列进行测序,确认了是丙酮丁醇梭菌(Clostridiumacetobutylicum)ATCC824株的adc序列(CA-P0165)。将这样得到的质粒命名为pT7Blue-ADC。pT7Blue-CTFA的制作在以下的条件下进行PCR。引物ctfA-F:5’-ATGAACTCTAAAATAATTAGATTTGAAAATTTAAGG-3’(序列号81)ctfA-R:5’-TTATGCAGGCTCCTTTACTATATAATTTA-3’(序列号82)反应条件模板:0.4μg的梭菌的基因组DNA引物:50pmol引物DNA,反应液:包含1xPfuUltraIIreactionbuffer(Stratagene社制)、10nmoldNTP、1μlPfuUltraIIfusionHSDNApolymerase(Stratagene社制)的50μl溶液反应:95℃5分钟-(95℃30秒、60℃30秒、72℃2分钟)×30个循环-72℃3分钟-4℃存放将扩增的657bp的片段使用Novagen社制PerfectlyBluntCloningKit同样地进行克隆。对克隆的序列进行测序,确认了是丙酮丁醇梭菌(Clostridiumacetobutylicum)ATCC824株的ctfA序列(CA-P0163)。将这样得到的质粒命名为pT7Blue-CTFA。pT7Blue-CTFB的制作在以下的条件下进行PCR。引物ctfB-F:5’-ATGATTAATGATAAAAACCTAGCGAAAG-3’(序列号83)ctfB-R:5’-CTAAACAGCCATGGGTCTAAGTTC-3’(序列号84)反应条件模板:0.4μg的梭菌的基因组DNA引物:50pmol引物DNA反应液:包含1xPfuUltraIIreactionbuffer(Stratagene社制)、10nmoldNTP、1μlPfuUltraIIfusionHSDNApolymerase(Stratagene社制)的50μl溶液反应:95℃5分钟-(95℃30秒、60℃30秒、72℃2分钟)×30个循环-72℃3分钟-4℃存放将扩增的666bp的片段使用Novagen社制PerfectlyBluntCloningKit进行克隆。对克隆的序列进行测序,确认了是丙酮丁醇梭菌(Clostridiumacetobutylicum)ATCC824株的ctfB序列(CA-P0164)。将这样得到的质粒命名为pT7Blue-CTFB。pDI626PGKpro的制作在以下的条件下进行PCR。引物SacI-Ppgk1FW:5’-TAGGGAGCTCCAAGAATTACTCGTGAGTAAGG-3’(序列号85)SacII-Ppgk1RV:5’-ATAACCGCGGTGTTTTATATTTGTTGTAAAAAGTAG-3’(序列号86)反应条件模板:0.4μg的酵母YPH499的基因组DNA引物:50pmol引物DNA,反应液:包含1xPfuUltraIIreactionbuffer(Stratagene社制)、10nmoldNTP、1μlPfuUltraIIfusionHSDNApolymerase(Stratagene社制)的50μl溶液反应:95℃5分钟-(95℃30秒、55℃30秒、72℃2分钟)×25个循环-72℃3分钟-4℃存放将反应液使用QIAGEN社制MinElutePCRpurificationkit进行纯化后,用限制性酶SacI和SacII消化。进行琼脂糖凝胶电泳,切下712bp的片段并使用QIAGEN社制MinEluteGelextractionkit进行纯化。与同样地用限制性酶SacI和SacII消化后的pDI626GAP((APP.Env.Micro.,2009,5536))载体连接。对所得的序列进行测序,确认制作了目的质粒。将这样得到的质粒命名为pDI626PGKpro。pDI626PGK的制作在以下的条件下进行PCR。引物SalI-Tpgk1FW:5’-TTAAGTCGACATTGAATTGAATTGAAATCGATAGATC-3’(序列号87)KpnI-Tpgk1RV2:5’-TTAAGGTACCGCTTCAAGCTTACACAACAC-3’(序列号88)反应条件模板:0.4μg的酵母YPH499的基因组DNA引物:50pmol引物DNA,反应液:包含1xPfuUltraIIreactionbuffer(Stratagene社制)、10nmoldNTP、1μlPfuUltraIIfusionHSDNApolymerase(Stratagene社制)的50μl溶液反应:95℃5分钟-(95℃30秒、55℃30秒、72℃2分钟)×25个循环-72℃3分钟-4℃存放将反应液使用QIAGEN社制MinElutePCRpurificationkit纯化后,用限制性酶SalI和KpnI消化。进行琼脂糖凝胶电泳,切下330bp的片段并使用QIAGEN社制MinEluteGelextractionkit进行纯化。与用限制性酶SalI和KpnI消化后的pDI626PGKpro载体连接。对所得的序列进行测序,确认制作了目的质粒。将这样得到的质粒命名为pDI626PGK。pDI626PGK-T的制作将pDI626PGK用限制性酶SbfI消化,将反应液使用QIAGEN社制MinElutePCRpurificationkit进行纯化。然后,使用TaKaRaBIO社制Bluntingkit将末端平滑化,进而用限制性酶KpnI消化。进行琼脂糖凝胶电泳,切下3650bp的片段并使用QIAGEN社制MinEluteGelextractionkit进行纯化。将此作为用于连接的载体。接着将pRS524GAP(APP.Env.Micro.,2009,5536)用限制性酶PmaCI和KpnI消化。进行琼脂糖凝胶电泳,切下765bp的片段并使用QIAGEN社制MinEluteGelextractionkit进行纯化作为插入片段。进行它们的连接,对所得序列的连接点进行测序,确认制作了目的质粒。将这样得到的质粒命名为pDI626PGK-T。pCR2.1-iPDH的制作以登录于GenBank(http://www.neb.nih.gov/Genbank/index.html)的来自拜氏梭菌(Clostridiumbeijerinckii)NRRLB593的adh:NADP-dependentalcoholdehydrogenase(NADP-依赖的醇脱氢酶)基因序列为基础合成最适合酿酒酵母的密码子的DNA序列(Operon社)。载体部分是pCR2.1(Invitrogen社制)。此外,将合成的DNA序列示于序列号89,将由合成的DNA序列所含的编码区所编码的氨基酸序列示于序列号90。将该质粒命名为pCR2.1-iPDH。pDI626PGK-T-iPDH的制作将pCR2.1-iPDH用限制性酶SacII和SalI消化,切下1080bp的片段,与同样地用限制性酶SacII和SalI消化后的pDI626PGK-T载体连接。对所得的序列进行测序,确认制作了目的质粒。将这样得到的质粒命名为pDI626PGK-T-iPDH。pENT-ADC的制作以pDI626-ADC为模板,用以下的引物进行PCR。引物08-189-adc-attB1-Fw:5’-GGGGACAAGTTTGTACAAAAAAGCAGGCTCAGTTCGAGTTTATCATTATC-3’(序列号91)08-189-adc-attB4-Rv:5’-GGGGACAACTTTGTATAGAAAAGTTGGGTGGGCCGCAAATTAAAGCCTTC-3’(序列号92)将所得的1809bp的PCR产物通过gatewayBP反应导入供体载体pDONR221P1-P4。进行所得的克隆的测序,确认了插入片段的全碱基序列中没有突变位点。将这样得到的质粒命名为pENT-ADC。pENT-CTFA的制作以pDI626PGK-CTFA为模板,使用以下的引物进行PCR。08-189-ctfA-attB4r-Fw:5’-GGGGACAACTTTTCTATACAAAGTTGGCTTCAAGCTTACACAACACGG-3’(序列号93)08-189-ctfA-attB3r-Rv:5’-GGGGACAACTTTATTATACAAAGTTGTCAAGAATTACTCGTGAGTAAGG-3’(序列号94)将所得的1823bp的PCR产物通过gatewayBP反应导入供体载体pDONR221P4r-P3r中。进行所得的克隆的测序,确认了在插入片段的全碱基序列中没有突变位点。将这样得到的质粒命名为pENT-CTFA。pDI626-CTFB的制作将pT7Blue-CTFB用限制性酶BamHI和SalI消化,切下771bp的片段,与同样地用限制性酶BamHI和SalI消化的pDI626载体连接。对所得的序列进行测序,确认制作了目的质粒。将这样得到的质粒命名为pDI626-CTFB(+A)。为了修正引物中的突变位点,使用以下的引物在以下的条件下进行PCR。BamHI-ctfB-F:5’-TAGTGGATCCGATGATTAATGATAAAAACC-3’(序列号95)pDI626MCS-seqF:5’-CCTAGACTTCAGGTTGTCTAAC-3’(序列号96)反应条件模板:1ng的pDI626-CTFB(+A)引物:50pmol引物DNA反应液:包含1xPfuUltraIIreactionbuffer(Stratagene社制)、10nmoldNTP、1μlPfuUltraIIfusionHSDNApolymerase(Stratagene社制)的50μl溶液反应:95℃2分钟-(95℃30秒、55℃30秒、72℃1分钟)×20个循环-72℃3分钟-4℃存放将反应液使用QIAGEN社制MinElutePCRpurificationkit纯化后,用限制性酶BamHI和SalI消化。进行琼脂糖凝胶电泳,切下702bp的片段并使用QIAGEN社制MinEluteGelextractionkit进行纯化。与用限制性酶BamHI和SalI消化后的pDI626载体连接。对所得的序列进行测序,确认了突变位点被修正。将这样得到的质粒命名为pDI626-CTFB。pENT-CTFB的制作以pDI626-CTFB为模板,使用以下的引物进行PCR。08-189-ctfB-attB3-Fw:5’-GGGGACAACTTTGTATAATAAAGTTGGGCCGCAAATTAAAGCCTTC-3’(序列号97)08-189-ctfB-attB2-Rv:5’-GGGGACCACTTTGTACAAGAAAGCTGGGTACAGTTCGAGTTTATCATTATC-3’(序列号98)将所得的1737bp的PCR产物通过gatewayBP反应导入供体载体pDONR221P3-P2中。进行所得的克隆的测序,确认了在插入片段的全碱基序列中没有突变位点。将这样得到的质粒命名为pENT-CTFB。pDEST626(2008)的制作在以下的条件下进行PCR。引物SacI-convA-F:5’-TAGGGAGCTCATCACAAGTTTGTACAAAAAAGCTG-3’(序列号99)KpnI-convA-R:5’-TTAAGGTACCATCACCACTTTGTACAAGAAAGC-3’(序列号100)反应条件模板:0.5ng的RfA(Invitrogen社制;Gateway载体转化系统的阅读框盒A)引物:50pmol引物DNA反应液:包含1xPfuUltraIIreactionbuffer(Stratagene社制)、10nmoldNTP、1μlPfuUltraIIfusionHSDNApolymerase(Stratagene)的50μl溶液反应:95℃2分钟-(95℃30秒、55℃30秒、72℃1分30秒)×20个循环-72℃3分钟-4℃存放将反应液使用QIAGEN社制MinElutePCRpurificationkit纯化后,用限制性酶SacI和KpnI消化。进行琼脂糖凝胶电泳,切下1717bp的片段并使用QIAGEN社制MinEluteGelextractionkit纯化后,与用限制性酶SacI和KpnI消化后的pDI626GAP载体(APP.Env.Micro.,2009,5536)连接。对所得的序列进行测序,确认制作了目的质粒。pEXP(Ura)-ADC-CTFA-CTFB的制作将所得的3个入门克隆(pENT-ADC、pENT-CTFA、pENT-CTFB)通过GatewayLR反应插入表达载体pDEST626(2008)中。对于所得的克隆通过PCR确认插入片段大小,确认了正确地进行了重组。进行测序,确认了序列中无错误。将这样得到的质粒命名为pEXP(Ura)-ADC-CTFA-CTFB。#3-17株的制作将表达载体pEXP(Ura)-ADC-CTFA-CTFB用限制性酶AatII、BssHII切割、线性化,乙醇沉淀后溶解于0.1×TE缓冲液中,使用FrozenEZyeasttransformationkit(Zymoresearch社制)转化酿酒酵母YPH499(Stratagene社制)。将所得的克隆进行菌落PCR,在25个克隆中确认导入了adc、ctfA、ctfB基因。将丙酮生产量最多的株命名为#3-17。#15-10株的制作将作为期待由丙酮向异丙醇转换的合成基因的ipdh基因的表达载体pDI626PGK-T-iPDH用限制性酶AatII和BssHII切割、线性化,乙醇沉淀后溶解于0.1×TE缓冲液中,使用FrozenEZyeasttransformationkit(Zymoresearch社制)转化丙酮生产酵母#3-17。将所得的14个克隆进行菌落PCR,在13克隆中确认了导入了ipdh基因。将异丙醇生产量最多的株命名为#15-10。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1