一种泰拉霉素有关物质、其制备方法及应用与流程
本发明涉及一种泰拉霉素有关物质、其制备方法及应用。
背景技术:
泰拉霉素(Tulathromycin)是半合成大环内酯类抗生素,于2004年在美国和欧盟上市。该药主要用于牛和猪由敏感菌引起的呼吸系统感染性疾病及由牛莫拉氏菌引起牛传染性角膜结膜炎的防治,药效强于市场上广泛使用的大环内酯类抗生素泰乐菌素和替米考星,在畜禽生产中的使用前景广阔。
泰拉霉素是由异构体A、B(分子式C41H79N3O12,分子量806.09)按9:1组成的十五元环大环内酯类抗生素,两种异构体可通过C11和C13之间内酯键的形成和断裂进行转换。泰拉霉素结构中含有三个极性氨基基团,PKa值在8.6~9.6之间,属于三胺类大环内酯抗生素,不同于氮杂环内酯和酮内酯类抗生素。
为了确保动物源性食品的安全,须对动物专用药物质量进行严格控制,药物含量测定、未知杂质的结构鉴定和杂质限量是药物质量控制的有效方法,杂质分析是药物质量标准的重要内容。泰拉霉素由半合成过程生产,与合成药物相比可控性更低,因此杂质谱更为复杂且难以预测。现行欧洲药典(EP)和美国药典(USP)均将兽药收录其中,并对有关物质限量提出要求,欧洲药典还要求对特定杂质进行控制(英国药典按惯例收录了欧洲药典的全部专论,内容一般不作修改)。VICH(兽用药物注册技术要求国际协调会)指导原则要求兽医专用原料药的杂质报告限度为0.10%,鉴定限度为0.20%,控制限度为0.50%(该指导不包括半合成抗生素);EMA(欧洲药品管理局)要求半合成兽用药物的杂质限度应符合VICH指导原则的要求。
目前,国内外尚未见采用LC-MS法分离检测泰拉霉素有关物质的报道。因此,亟需降解泰拉霉素以制备降解的有关物质,建立泰拉霉素有关物质的 LC-MS分离、检测方法,并对泰拉霉素有关物质进行鉴定。
技术实现要素:
本发明所要解决的问题是为了克服现有技术中缺乏泰拉霉素在合成及降解过程中产生的有关物质的分离、检测方法,且不能对之进行合成、鉴定、确证结构等缺陷,而提供了一种泰拉霉素有关物质、其制备方法及应用。本发明的泰拉霉素有关物质是对泰拉霉素进行质量控制的必需品;本发明的制备方法能够制得并有效分离泰拉霉素有关物质2,从而控制泰拉霉素的药品质量,为泰拉霉素未知杂质的研究奠定了良好的基础。
本发明提供了一种泰拉霉素有关物质的分离方法,其包括下述步骤:采用高效液相色谱法,将待测物在色谱柱中进行洗脱,即可;所述的待测物为泰拉霉素原料药或泰拉霉素降解物;所述的色谱柱为C18分析柱或C18制备柱;所述的洗脱的流动相A为用氨水调节pH值为7~8的、体积分数为0.1%~0.4%甲酸水溶液,所述的洗脱的流动相B为甲醇与乙腈的体积比为(1.5~2.0):1的混合溶剂;
当所述的色谱柱为C18分析柱时,所述的洗脱的参数如下:0min→15min,A:B=(60~70):(40~30),15min→40min,A:B=(60~70):(40~30)→(25~35):(75~65),40→55min,A:B=(25~35):(75~65);所述的A:B指所述的流动相A与所述的流动相B的体积比;
当所述的色谱柱为C18制备柱时,所述的洗脱的参数如下:所述的流动相A与所述的流动相B的体积比为(75~85):(25~15)。
在所述的分离方法中,所述的泰拉霉素原料药可为本领域常规的泰拉霉素原料药,较佳地为江苏凌云药业有限公司生产的泰拉霉素原料药。
在所述的分离方法中,所述的泰拉霉素降解物为泰拉霉素经降解反应得到的物质;所述的降解反应可为本领域常规的降解反应,较佳地为酸降解反应、碱降解反应、高温降解反应、高湿降解反应、氧化降解反应或光照降解反应;
所述的氧化降解反应可包括下述步骤:在乙腈中,将泰拉霉素与双氧水水溶液进行氧化降解反应,得到即可;
所述的碱降解反应可包括下述步骤:在乙腈中,将泰拉霉素与氢氧化钠水溶液进行碱降解反应,得到即可。
在所述的分离方法中,所述的待测物可采用本领域常规的方法进样,较佳地以待测物的乙腈溶液的形式进样;当所述的待测物为泰拉霉素经氧化降解反应得到的物质时,较佳地,所述的氧化降解反应的反应液经乙腈稀释后形成所述的待测物的乙腈溶液;当所述的待测物为泰拉霉素经碱降解反应得到的物质时,较佳地,所述的碱降解反应的反应液经酸中和、并用乙腈稀释后形成所述的待测物的乙腈溶液;当所述的待测物为泰拉霉素经酸降解反应得到的物质时,较佳地,所述的酸降解反应的反应液经碱中和、并用乙腈稀释后形成所述的待测物的乙腈溶液;
所述的酸中和的酸可为本领域常规的酸,较佳地为盐酸水溶液,更佳地为0.1mol/L的盐酸水溶液;所述的碱中和的碱可为本领域常规的碱,较佳地为NaOH水溶液,更佳地为0.1mol/L的NaOH水溶液;
所述的待测物的乙腈溶液的浓度可为本领域常规的浓度,较佳地为3g/L~50g/L,更佳地为5g/L~20g/L;
所述的待测物的乙腈溶液的进样量可为本领域常规的进样量,当所述的 色谱柱为C18分析柱时,所述的进样量较佳地为5μL~100μL,更佳地为20μL~50μL;当所述的色谱柱为C18制备柱时,所述的进样量较佳地为50μL~500μL,更佳地为50μL~300μL。
在所述的分离方法中,所述的C18分析柱可为本领域常规的C18分析柱,较佳地为XbridgeTM C18(250×4.6mm,5μm)分析柱;所述的C18制备柱可为本领域常规的C18制备柱,较佳地为XbridgeTM C18(19*50mm,5μm)制备柱。
在所述的分离方法中,所述的流动相B中甲醇与乙腈的体积比较佳地为45:25;当所述的色谱柱为C18分析柱时,所述的流动相A的pH值较佳地为7.6~7.99(例如7.66),所述的流动相A的甲酸水溶液的体积分数较佳地为0.3%~0.35%,更佳地,为用氨水调节pH值为7.6的、体积分数为0.35%的甲酸水溶液;当所述的色谱柱为C18制备柱时,所述的流动相A的pH值较佳地为7.6~7.8,所述的流动相A的甲酸水溶液的体积分数较佳地为0.3%~0.35%。
在所述的分离方法中,当所述的色谱柱为C18分析柱时,所述的洗脱的参数较佳地如下:0min→15min,A:B=65:35,15min→40min,A:B=65:35→30:70,40→55min,A:B=30:70;当所述的色谱柱为C18制备柱时,所述的洗脱的参数较佳地如下:所述的流动相A与所述的流动相B的体积比为80:20。
在所述的分离方法中,所述的氨水为本领域常规的氨水,较佳地为质量分数为25%~28%的氨水。
在所述的分离方法中,可采用本领域常规的高效液相色谱,当所述的色谱柱为C18分析柱时,较佳地采用Waters公司的Alliance 2695/ZQ液相色谱-质谱联用仪;当所述的色谱柱为C18制备柱时,较佳地采用Waters公司的2767型自动纯化仪。
在所述的分离方法中,所述的分离方法的流速可为本领域常规的流速;当所述的色谱柱为C18分析柱时,所述的流速较佳地为 0.7ml/min~1.3ml/min,0.8ml/min~1.2ml/min,例如1.0ml/min;当所述的色谱柱为C18制备柱时,所述的流速较佳地为10ml/min~25ml/min,更佳地为15ml/min~20ml/min,例如17ml/min。
在所述的分离方法中,所述的流动相A和所述的流动相B可采用本领域常规的处理方法进行预处理,较佳地经0.22μm滤膜过滤,并超声10min。
在所述的分离方法中,所述的分离方法的柱温可为本领域常规的柱温,较佳地为20℃~40℃,更佳地为25℃~35℃,例如30℃。
在所述的分离方法中,所述的分离方法检测时的紫外吸收波长可为本领域常规的紫外吸收波长,较佳地为200~215nm,更佳地为205~210nm;经过紫外检测器后的流份的1/5~1/3进入质谱仪检测。
本发明还提供了一种泰拉霉素有关物质1或其盐,
本发明还提供了所述的泰拉霉素有关物质1的富集制备方法,其如上述的泰拉霉素有关物质的分离方法,收集所述的泰拉霉素有关物质1即可。
本发明还提供了所述的泰拉霉素有关物质1或其盐在泰拉霉素质量控制中作为杂质鉴定的应用。
本发明还提供了一种泰拉霉素有关物质2或其盐,
本发明还提供了所述的泰拉霉素有关物质2的制备方法,其包括下述步骤:在乙腈中,将泰拉霉素与双氧水水溶液进行氧化降解反应,得到所述的泰拉霉素有关物质2即可。
在所述的氧化降解反应中,所述的泰拉霉素与所述的乙腈的质量体积比可为本领域中氧化降解反应的常规质量体积比,较佳地为3g/L~50g/L,更佳地为5g/L~20g/L。
在所述的氧化降解反应中,所述的双氧水水溶液可为本领域常规的双氧水水溶液,较佳地为质量分数为0.1%~30%的双氧水水溶液,更佳地为0.15%~0.5%的双氧水水溶液。
在所述的氧化降解反应中,所述的泰拉霉素与所述的双氧水水溶液的质量体积比可为本领域中氧化降解反应的常规质量体积比,较佳地为40g/L~90g/L,更佳地为50g/L~75g/L。
在所述的氧化降解反应中,所述的氧化降解反应的温度可为本领域中氧化降解反应的常规温度,较佳地为20℃~50℃,更佳地为20℃~30℃。
在所述的氧化降解反应中,所述的氧化降解反应的进程可采用本领域中的常规监测方法(例如LC-MS)进行监测,所述的降解反应的时间较佳地为1min~30min,更佳地为5min~20min。
所述的氧化降解反应较佳地还包括后处理;所述的后处理可为本领域中氧化降解反应的常规后处理,较佳地,所述的氧化降解反应的反应液按照上述的泰拉霉素有关物质的分离方法进行后处理。
本发明还提供了所述的泰拉霉素有关物质2或其盐在泰拉霉素质量控制中作为杂质鉴定的应用。
本发明提供了一种泰拉霉素有关物质3或其盐,
本发明还提供了所述的泰拉霉素有关物质3的富集制备方法,其如上述的泰拉霉素有关物质的分离方法,收集所述的泰拉霉素有关物质3即可。
本发明还提供了所述的泰拉霉素有关物质3或其盐在泰拉霉素质量控制中作为杂质鉴定的应用。
本发明还提供了一种泰拉霉素有关物质4或其盐,
本发明还提供了所述的泰拉霉素有关物质4的制备方法,其包括下述步骤:在乙腈中,将泰拉霉素与氢氧化钠水溶液进行碱降解反应,得到所述的泰拉霉素有关物质4即可。
在所述的碱降解反应中,所述的泰拉霉素与所述的乙腈的质量体积比可为本领域中碱降解反应的常规质量体积比,较佳地为3g/L~50g/L,更佳地为5g/L~20g/L。
在所述的碱降解反应中,所述的氢氧化钠水溶液可为本领域常规的氢氧化钠水溶液,较佳地为0.1mol/L~10mol/L的氢氧化钠水溶液,更佳地为1mol/L~5mol/L的氢氧化钠水溶液。
在所述的碱降解反应中,所述的泰拉霉素与所述的氢氧化钠水溶液的质量体积比可为本领域中碱降解反应的常规质量体积比,较佳地为 40g/L~90g/L,更佳地为50g/L~75g/L。
在所述的碱降解反应中,所述的碱降解反应的温度可为本领域中碱降解反应的常规温度,较佳地为50℃~90℃,更佳地为55℃~70℃,例如60℃。
在所述的碱降解反应中,所述的碱降解反应的进程可采用本领域中的常规监测方法(例如TLC、HPLC或LC-MS)进行监测,所述的碱降解反应的时间较佳地为15min~50min,更佳地为20min~40min,例如25min。
所述的碱降解反应较佳地还包括后处理;所述的后处理可为本领域中碱降解反应的常规后处理,较佳地,所述的碱降解反应的反应液按照上述的泰拉霉素有关物质的分离方法进行后处理。
本发明还提供了所述的泰拉霉素有关物质4或其盐在泰拉霉素质量控制中作为杂质鉴定的应用。
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
本发明所用试剂和原料均市售可得。
本发明的积极进步效果在于:本发明的泰拉霉素有关物质是对泰拉霉素进行质量控制的必需品;本发明的制备方法能够制得并有效分离泰拉霉素有关物质2,从而控制泰拉霉素的药品质量,为泰拉霉素未知杂质的研究奠定了良好的基础。
附图说明
图1为实施例1得到的泰拉霉素原料药质谱总离子流色谱图。
图2为有关物质1的一级质谱图。
图3为有关物质1的二级质谱图。
图4为有关物质3的一级质谱图。
图5为有关物质3的二级质谱图。
图6为实施例2得到的泰拉霉素原料药质谱总离子流色谱图。
图7为实施例3得到的泰拉霉素原料药质谱总离子流色谱图。
图8为实施例4得到的泰拉霉素原料药质谱总离子流色谱图。
图9为实施例5得到的泰拉霉素原料药质谱总离子流色谱图。
图10为实施例6得到的泰拉霉素原料药质谱总离子流色谱图。
图11为实施例7得到的泰拉霉素原料药质谱总离子流色谱图。
图12为实施例8得到的泰拉霉素原料药质谱总离子流色谱图。
图13为实施例9得到的泰拉霉素原料药质谱总离子流色谱图。
图14为实施例10得到的泰拉霉素原料药质谱总离子流色谱图。
图15为泰拉霉素原料药氧化降解反应后,经分析柱的质谱总离子流色谱图。
图16为泰拉霉素原料药氧化降解反应后,经制备柱的质谱总离子流色谱图。
图17为有关物质2的一级质谱图。
图18为有关物质2的二级质谱图。
图19为泰拉霉素原料药碱降解反应后,经分析柱的质谱总离子流色谱图。
图20为有关物质4的一级质谱图。
图21为有关物质4的二级质谱图。
图22为泰拉霉素原料药酸降解反应后,经分析柱的质谱总离子流色谱图。
图23为对比例1得到的泰拉霉素原料药质谱总离子流色谱图。
图24为对比例2得到的泰拉霉素原料药质谱总离子流色谱图。
图25为对比例3得到的泰拉霉素原料药质谱总离子流色谱图。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
本发明所使用试剂如下:
乙腈、甲醇为色谱纯(Thermo Fisher Scientific公司);甲酸(98%)、氨水(25wt%~28wt%)、30wt%过氧化氢、盐酸为分析纯,氢氧化钠(国药集团化学制剂有限公司);水为娃哈哈纯净水(过0.22μm水膜);泰拉霉素原料药由江苏凌云药业有限公司提供。
本发明所使用仪器如下:
(1)2695型高效液相色谱仪;2487型紫外检测器;Micromass ZQ质谱仪;Q-Tof micro质谱仪;Masslynx色谱工作站(Waters公司)
(2)Q-Exactive四级杆轨道阱高分辨质谱(Thermo公司)
(3)2767型自动纯化仪
本发明所使用的质谱方法如下:
Micromass ZQ:检测模式ESI(+);喷雾电压3kV;锥孔电压30V;源温100℃;脱溶剂温度250℃;全扫描范围m/z100~1500。
Q-Exactive四级杆轨道阱高分辨质谱参数:HESI喷雾电压:+3.0KV/-2.7KV(正负切换同时扫描);鞘气压力:35arb;辅助气压力:10arb;毛细管温度:300℃;加热温度:300℃;扫描模式:Full MS(分辨率7000)和dd-MS2(分辨率17500)
实施例1
5mg泰拉霉素原料药溶于1mL乙腈,进样量20μL。
色谱参数:XbridgeTM C18(250*4.6mm,5μm)柱,流动相A:0.35%甲酸水溶液(氨水调节pH值7.66),流动相B:甲醇:乙腈=45:25,梯度洗脱0→15min,A:B=65:35;15→40min,A:B=65:35→30:70;40→55min,A:B=30:70,紫外吸收波长205nm,柱温35℃,流速1.0ml/min,HPLC流份经UV检测器后以3:1分流进入MS检测。质谱总离子流如图1所示,各杂质如表1所示。流动相盐浓度为0.35%时,主峰保留时间合适,峰形良好。有关物质与主峰能良好分离。从图中可以看出,本发明的方法可用于检测泰拉霉素及其杂质,该方法可应用于泰拉霉素原料药合成工艺监控及质量控 制。
表1图1中的各杂质
注:Aera%为面积归一化法测得百分含量。
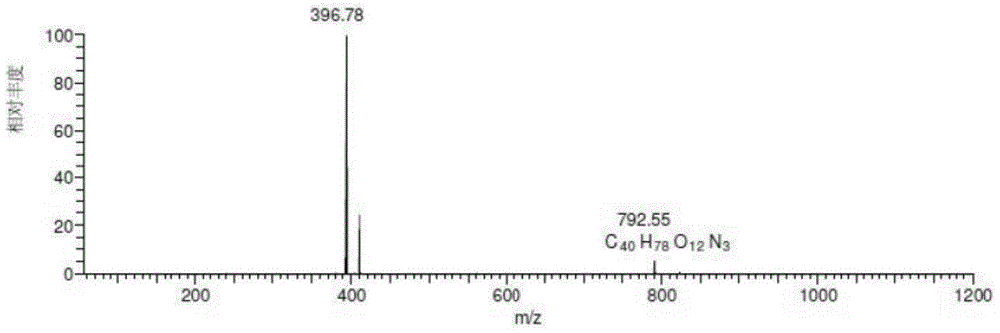
表1中的4即本发明的有关物质1,高分辨测得分子式为C40H77N3O12([M+H]+=792.55829),MS2碎片为689.45898(C35H63NO12)、563.39099(C28H55N2O9)、420.29337(C21H42NO7)、230.17519(C12H24NO3)。其一级质谱图如图2所示,二级质谱图如图3所示,质谱裂解途径如下所示:
表1中的13即本发明的有关物质3,高分辨测得分子式为C39H72N2O12([M+H]+=761.51624),MS2碎片为532.34827(C27H50NO9)、230.17520(C12H24NO3)。其一级质谱图如图4所示,二级质谱图如图5所示,质谱裂解途径如下所示:
实施例2
5mg泰拉霉素原料药溶于1mL乙腈,进样量20μL。
色谱参数:XbridgeTM C18(250*4.6mm,5μm)柱,流动相A:0.3%甲 酸水溶液(氨水调节pH值7.66),流动相B:甲醇:乙腈=45:25,梯度洗脱0→15min,A:B=65:35;15→40min,A:B=65:35→30:70;40→55min,A:B=30:70,紫外吸收波长205nm,柱温35℃,流速1.0ml/min,HPLC流份经UV检测器后以3:1分流进入MS检测。质谱总离子流如图6所示。流动相盐浓度为0.3%时,主峰保留时间合适,峰形良好。有关物质与主峰能良好分离。
实施例3
5mg泰拉霉素原料药溶于1mL乙腈,进样量20μL。
色谱参数:XbridgeTM C18(250*4.6mm,5μm)柱,流动相A:0.4%甲酸水溶液(氨水调节pH值7.66),流动相B:甲醇:乙腈=45:25,梯度洗脱0→15min,A:B=65:35;15→40min,A:B=65:35→30:70;40→55min,A:B=30:70,紫外吸收波长205nm,柱温35℃,流速1.0ml/min,HPLC流份经UV检测器后以3:1分流进入MS检测。质谱总离子流如图7所示。流动相盐浓度为0.4%时,主峰保留时间合适,峰形良好。有关物质与主峰能良好分离。
实施例4
5mg泰拉霉素原料药溶于1mL乙腈,进样量20μL。
色谱参数:XbridgeTM C18(250*4.6mm,5μm)柱,流动相A:0.1%甲酸水溶液(氨水调节pH值7.99),流动相B:甲醇:乙腈=45:25,梯度洗脱0→15min,A:B=65:35;15→40min,A:B=65:35→30:70;40→55min,A:B=30:70,紫外吸收波长205nm,柱温35℃,流速1.0ml/min,HPLC流份经UV检测器后以3:1分流进入MS检测。质谱总离子流如图8所示。流动相盐浓度为0.1%时,pH值为7.99时,主峰保留时间合适,峰形对称。有关物质与主峰能良好分离。
实施例5
5mg泰拉霉素原料药溶于1mL乙腈,进样量20μL。
色谱参数:XbridgeTM C18(250*4.6mm,5μm)柱,流动相A:0.35%甲 酸水溶液(氨水调节pH值7.6),流动相B:甲醇:乙腈=1.5:1,梯度洗脱0→15min,A:B=65:35;15→40min,A:B=65:35→30:70;40→55min,A:B=30:70,紫外吸收波长205nm,柱温35℃,流速1.0ml/min,HPLC流份经UV检测器后以3:1分流进入MS检测。质谱总离子流如图9所示。流动相B中甲醇:乙腈=1.5:1时,主峰保留时间合适,峰形对称。有关物质与主峰能良好分离。
实施例6
5mg泰拉霉素原料药溶于1mL乙腈,进样量20μL。
色谱参数:XbridgeTM C18(250*4.6mm,5μm)柱,流动相A:0.35%甲酸水溶液(氨水调节pH值7.6),流动相B:甲醇:乙腈=45:25,梯度洗脱0→15min,A:B=65:35;15→40min,A:B=65:35→35:65;40→55min,A:B=35:65,紫外吸收波长205nm,柱温35℃,流速1.0ml/min,HPLC流份经UV检测器后以3:1分流进入MS检测。质谱总离子流如图10所示。流动相梯度洗脱程序为0→15min,A:B=65:35;15→40min,A:B=65:35→35:65;40→55min,A:B=35:65时,主峰保留时间合适,峰形对称。有关物质与主峰能良好分离。
实施例7
5mg泰拉霉素原料药溶于1mL乙腈,进样量20μL。
色谱参数:XbridgeTM C18(250*4.6mm,5μm)柱,流动相A:0.35%甲酸水溶液(氨水调节pH值7.6),流动相B:甲醇:乙腈=45:25,梯度洗脱0→15min,A:B=70:30;15→40min,A:B=70:30→30:70;40→55min,A:B=30:70,紫外吸收波长205nm,柱温35℃,流速1.0ml/min,HPLC流份经UV检测器后以3:1分流进入MS检测。质谱总离子流如图11所示。流动相梯度洗脱程序为0→15min,A:B=70:30;15→40min,A:B=70:30→30:70;40→55min,A:B=30:70时,主峰保留时间合适,峰形对称。有关物质与主峰能良好分离。
实施例8
5mg泰拉霉素原料药溶于1mL乙腈,进样量20μL。
色谱参数:XbridgeTM C18(250*4.6mm,5μm)柱,流动相A:0.35%甲酸水溶液(氨水调节pH值7.6),流动相B:甲醇:乙腈=2:1,梯度洗脱0→15min,A:B=70:30;15→40min,A:B=70:30→30:70;40→55min,A:B=30:70,紫外吸收波长205nm,柱温35℃,流速1.2ml/min,HPLC流份经UV检测器后以3:1分流进入MS检测。质谱总离子流如图12所示。流动相B为甲醇:乙腈=2:1,流速1.2ml/min时,主峰保留时间合适,峰形对称。有关物质与主峰能良好分离。
实施例9
5mg泰拉霉素原料药溶于1mL乙腈,进样量20μL。
色谱参数:XbridgeTM C18(250*4.6mm,5μm)柱,流动相A:0.35%甲酸水溶液(氨水调节pH值7.0),流动相B:甲醇:乙腈=45:25,梯度洗脱0→15min,A:B=70:30;15→40min,A:B=70:30→35:65;40→55min,A:B=35:65,紫外吸收波长205nm,柱温35℃,流速1.0ml/min,HPLC流份经UV检测器后以3:1分流进入MS检测。质谱总离子流如图13所示。流动相A的pH值7.0,梯度为0→15min,A:B=70:30;15→40min,A:B=70:30→35:65;40→55min,A:B=35:65时,主峰保留时间合适,峰形对称。有关物质与主峰能良好分离。
实施例10
5mg泰拉霉素原料药溶于1mL乙腈,进样量20μL。
色谱参数:XbridgeTM C18(250*4.6mm,5μm)柱,流动相A:0.35%甲酸水溶液(氨水调节pH值8.0),流动相B:甲醇:乙腈=45:25,梯度洗脱0→15min,A:B=60:40;15→40min,A:B=60:40→25:75;40→55min,A:B=25:75,紫外吸收波长205nm,柱温35℃,流速1.0ml/min,HPLC流份经UV检测器后以3:1分流进入MS检测。质谱总离子流如图14所示。流动相A的pH值8.0,梯度为0→15min,A:B=60:40;15→40min,A:B=60:40→25:75;40→55min,A:B=25:75时,主峰保留时间合适,峰形对称。有关物 质与主峰能良好分离。
实施例11氧化降解反应
取约50mg泰拉霉素原料药于10ml量瓶中,加0.15%H2O2溶液1ml,加1ml乙腈助溶,室温(20℃)放置5min,乙腈定容,0.22μm滤膜过滤,按实施例1的色谱参数测定,进样20μl。质谱总离子流图如图15所示。
反应液还可按照下述色谱条件富集:XBridge C18(19*50mm,5μm)制备柱;进样500μL;流动相A:0.35%甲酸水溶液(氨水调节pH值为7.8);流动相B:甲醇:乙腈=45:25;A:B=80:20;紫外吸收波长:205nm;流速:17ml/min,质谱总离子流图如图16所示。
图15中23.32min处的化合物即本发明的有关物质2,高分辨测得分子式为C41H79N3O13([M+H]+=822.56921),比泰拉霉素分子式中多了一个O。MS2碎片761.35416(C39H73N2O12+)、593.40082(C29H57N2O10)、532.34883(C27H50NO9+)、230.17529(C12H24NO3+)。其一级质谱图如图17所示,二级质谱图如图18所示,质谱裂解途径如下所示:
实施例12碱降解反应
取约50mg泰拉霉素原料药于10ml量瓶中,加1M NaOH溶液1ml,加1ml乙腈助溶,60℃水浴加热25min,0.1M HCl溶液中和,乙腈定容,0.22μm滤膜过滤,按实施例1的色谱参数测定,进样20μl。质谱总离子流图如图 19所示。
图19中的7.80min处的化合物即本发明的有关物质4,高分辨测得分子式为C41H81N3O13([M+H]+=824.58527),MS2碎片为595.41656(C29H59N2O10)、550.38651(C27H52NO10+)、420.29623(C21H42NO7)、402.28582(C21H40NO6)、158.11700(C8H16NO2+)、230.17590(C12H24NO3+)。其一级质谱图如图20所示,二级质谱图如图21所示,质谱裂解途径如下所示:
实施例13酸降解反应
取约50mg泰拉霉素样品于10ml量瓶中,加1M HCL溶液1ml,室温(20℃)放置4h,1M NaOH溶液中和,乙腈定容,0.22μm滤膜过滤,按实施例1的色谱参数测定,进样20μl。质谱总离子流图如图22所示。
实施例14高温降解反应
条件1取泰拉霉素原料药适量,置于105℃烘箱中24h,放冷,取5mg溶于1ml乙腈中,0.22μm滤膜过滤后,按实施例1的色谱参数测定,进样20μL。
条件2取约50mg泰拉霉素样品于10ml量瓶中,乙腈定容,置沸水浴中12h,放冷,0.22μm滤膜过滤,按实施例1的色谱参数测定,进样20μl。
高温破坏分析:泰拉霉素固体样品高温破坏与原料药相比,未见新的杂质峰出现,样品在说明该样品在高温下较稳定。
实施例15光照降解反应
取泰拉霉素适量,置于光照箱(照度45Lx±500Lx)光照10天,分别于第5天和第10天取样,按实施例1的色谱参数测定,结果与未光照处理的样品对比发现样品光照5天后分子量为818(即表1中的11)的杂质含量增大,光照10天后此杂质含量增大。
实施例16高湿降解反应
在密闭容器下部放置KNO3饱和溶液(25℃,湿度92.5%),取泰拉霉素适量,在该容器中放置10天,于第5天和第10天取样,按实施例1的色谱参数测定,结果与未高湿处理的样品比较,未见新的杂质峰出现,说明样品受湿度影响不大。
泰拉霉素在高温、高湿、光照、酸降解反应中未发现新的有关物质。
对比例1流动相A为醋酸铵
色谱条件:XbridgeTM C18(250*4.6mm,5μm)柱;流动相A:15mM醋酸铵,氨水调节pH值为7.8;流动相B:甲醇:乙腈=45:25;梯度洗脱0→15min,A:B=65:35;15→40min,A:B=65:35→30:70;40→55min,A:B=30:70;紫外吸收波长205nm,柱温35℃,流速1.0ml/min;除上述参数外,其余参数均同实施例1,质谱总离子流图如图23所示,其保留时间长,杂质分离差。
对比例2流动相B为乙腈
色谱条件:XbridgeTM C18(250*4.6mm,5μm)柱;流动相A:0.35%甲酸(氨水调节pH值7.66);流动相B:乙腈;梯度洗脱0→15min,A:B=65:35;15→40min,A:B=65:35→30:70;40→55min,A:B=30:70;紫外吸收波长205nm,柱温35℃,流速1.0ml/min;除上述参数外,其余参数均同实施例1,质谱总离子流图如图24所示,其保留时间太短,杂质分离差。
对比例3流动相B为甲醇:乙腈=1:1
色谱条件:XbridgeTM C18(250*4.6mm,5μm)柱;流动相A:0.35%甲酸(氨水调节pH值7.66);流动相B:甲醇:乙腈=1:1;梯度洗脱0→15min,A:B=65:35;15→40min,A:B=65:35→30:70;40→55min,A: B=30:70;紫外吸收波长205nm,柱温35℃,流速1.0ml/min;除上述参数外,其余参数均同实施例1,质谱总离子流图如图25所示,其杂质分离差。
- 还没有人留言评论。精彩留言会获得点赞!