一种α‑酮基丁酸的发酵生产工艺的制作方法
本发明涉及化合物生物技术生产领域,尤其是一种α-酮基丁酸的发酵生产工艺。
背景技术:
α-酮基丁酸的生产方法包括化学合成法和生物合成法。化学法是由草酸二乙酯和丙酸乙酯混合水解得到α-酮基丁酸。生物法合成α-酮基丁酸主要为前体物转化法和直接发酵法。Nakahara等(1994)以1,2-丁二醇为碳源和底物,采用马红球菌(Rhodococcus equi)IF03730将其转化为α-酮基丁酸,培养32h转化率为68.2%。随后研究发现恶臭假单胞菌(Pseudomonas sputita)和施氏假单胞菌(Pseudomonas stutzeri)SDM分别能将巴豆酸和DL-2-羟基丁酸转化为α-酮基丁酸,其产量为4.8g/L。马翠卿等(ZL201110376233.8)报道了以L-苏氨酸为底物利用含L-苏氨酸脱水酶的假单胞菌、棒杆菌属或芽孢杆菌属为生物催化剂制备α-酮基丁酸的方法,转化液中α-酮基丁酸的浓度为25.6g/L,转化率为99.6%。陈宁等(201410132996.1)构建了过表达的苏氨酸脱水酶编码基因ilvA、敲除乙酰羟基酸合成酶Ⅰ大亚基编码基因ilvB、乙酰羟基酸合成酶Ⅲ大亚基编码基因ilvI以及苏氨酸操纵子前导肽编码基因thrL的基因工程大肠杆菌,该菌株以葡萄糖为底物发酵20-24h后,α-酮基丁酸最高产量达到15.7g/L。该方法通过低拷贝质粒pwsk29直接过表达苏氨酸脱水酶编码基因ilvA,且出发菌株为大肠杆菌E.col iMG1655标准株。
目前工业生产α-酮基丁酸主要采用化学合成法,但该方法存在反应条件复杂、能耗大、污染重等问题。而生物法的前体物转化法存在着底物生产成本高,微生物制剂难于培养等问题;生物法中利用重组大肠杆菌直接发酵生产α-酮基丁酸最大的问题在于产物毒性高,如图1所示,α-酮基丁酸浓度达到8g/L以上时,α-酮基丁酸对大肠杆菌菌体生长和产物合成存在严重的抑制作用。同时通过实验发现,敲除乙酰羟基酸合成酶Ⅰ基因ilvBN会影响缬氨酸的合成,菌体生物量大幅度下降,且对提升α-酮基丁酸的积累量无明显作用。
技术实现要素:
本发明所要解决的技术问题在于提供一种α-酮基丁酸的发酵生产工艺,可高效制备α-酮基丁酸。
为解决上述技术问题,本发明的技术方案是:
一种α-酮基丁酸的发酵生产工艺,对大肠杆菌E.col iTHRD(保藏号CGMCC No.11074)苏氨酸脱水酶编码基因ilvA进行定点突变、解除L-异亮氨酸对ilvA的反馈抑制的基础上过表达ilvA;敲除了乙酰羟基酸合成酶III的大亚基编码基因ilvI;利用温度控制诱导的方式,采用发酵法生产α-酮丁酸,其中,最佳诱导时机和最合适的升温诱导模式为:35-37℃培养菌体,生长到OD600为10-15时,1h内升温至40-42℃(快速升温)。
优选的,上述α-酮基丁酸的发酵生产工艺,利用温控诱导方式制备α-酮基丁酸,具体步骤如下:
Ⅰ、摇瓶发酵积累α-酮基丁酸
<1>活化斜面培养:将产生α-酮基丁酸的基因工程菌划线接于1支新鲜AmpR抗性斜面,37℃培养15-17h;
<2>摇瓶种子培养:从新鲜活化斜面上挑取1环菌体接于30mL的种子培养基中,200r/min,35-37℃恒温振荡培养,种子培养期间根据指示剂苯酚红颜色变化,用25%(W/V)氨水调节pH≈6.7-7.0;
<3>摇瓶发酵培养:保证溶氧充足情况下,500mL挡板三角瓶装液量30mL,9层纱布封口;待种子液培养至OD600值约为8-10时,种龄为10-12h,按8-10%接种量接入发酵培养基中,200r/min,32-35℃恒温振荡培养;待发酵4-6h,菌体OD600值约为10-15时,将发酵温度从32-35℃直接提高到40-42℃开始升温诱导,直至24h发酵结束,发酵期间补加25%(W/V)的氨水维持发酵液pH≈6.7-7.0;
上述步骤(3)所得α-酮基丁酸的产量可达到9.4-12.5g/L;
或
Ⅱ、7.5L发酵罐发酵积累α-酮基丁酸
<1>活化斜面培养:将产生α-酮基丁酸的基因工程菌划线接于4支新鲜AmpR抗性斜面,37℃培养16h左右;
<2>7.5L罐种子培养:吸取适量灭菌的去离子水于4支新鲜的活化斜面,刮下所有菌体后将菌悬液接入装有2L种子培养基的5L发酵罐;种子培养温度为35-37℃,通气量变化范围为2-5L/min,搅拌转速变化范围为100-500r/min,期间通过流加25%(W/V)的氨水控制pH≈6.7-7.0;
<3>7.5L罐发酵培养:种子液培养至OD600=10-15时,种龄约为7h,将500mL左右种子液转移到含有3.5L新鲜无菌发酵培养基的7.5L发酵罐中,总装液量为4L,接种量为8-10%;32-35℃开始进行发酵培养;待发酵液OD600值为20-25时,在0.5-1.0h内将发酵温度从32-35℃提高到40-42℃开始升温诱导,直至28h发酵结束,发酵过程中通过自动流加25%(W/V)氨水控制pH≈6.7-7.0,溶氧控制在30-50%,通气量变化范围为2-8L/min,搅拌转速变化范围为200-900r/min;当发酵培养基中葡萄糖耗完后,将80%(W/V)葡萄糖溶液按1-1.5mL/min的脉冲速度流加至培养基中以维持发酵液中残糖浓度控制在0-1g/L;
上述步骤(3)所得α-酮基丁酸的产量可达到16.5-22.8g/L。
上述α-酮基丁酸的制备方法,所得发酵液中α-酮基丁酸的检测方法为:发酵液样品经13000转离心2min,上清液经0.22μm膜过滤后用UltiMate3000(Thermo Scientific)高效液相测试α-酮丁酸产量,检测条件各参数为:色谱柱aminexR HPX-87H(300mm×7.8mm),流动相为5mmol/L H2SO4,流速0.5mL/min,柱温30℃,检测波长215nm,进样量为20μL。
优选的,上述α-酮基丁酸的发酵生产工艺,所述产生α-酮基丁酸的基因工程菌是以苏氨酸生产菌株E.coli THRD(保藏号CGMCC No.11074)为出发菌株,利用热诱导型表达载体PBV220、PBV221或PBV222过表达定点突变的苏氨酸脱水酶编码基因ilvA并敲除乙酰羟基酸合成酶III的大亚基编码基因ilvI。
优选的,上述α-酮基丁酸的发酵生产工艺,所述定点突变的苏氨酸脱水酶编码基因ilvA的核苷酸序列为序列表<400>2所示序列。
所述编码基因ilvA原始(未突变)核苷酸序列为序列表<400>1所示序列;所述编码基因ilvI的核苷酸序列为序列表<400>3所示序列;
优选的,上述α-酮基丁酸的发酵生产工艺,所述产生α-酮基丁酸的基因工程菌是由下述方法构建得到的,具体步骤为:
一、基因ilvI的敲除
<1>采用PCR技术以大肠杆菌E.coli THRD(保藏号CGMCC No.11074)基因组为模板,根据E.col iMG1655中ilvI(GeneID:948793)基因的5’和3’端500bp序列设计同源臂引物,扩增获取ilvI基因的上下游同源臂;
<2>采用PCR技术以pKD3质粒为模板,设计引物,扩增氯霉素抗性基因片段;
<3>以<1>、<2>获得的扩增片段为模板通过重叠PCR获得ilvI基因的敲除片段,所述敲除片段由乙酰羟基酸合成酶III大亚基编码基因ilvI的上、下游同源臂基因片段以及氯霉素抗性基因片段组成;
<4>将上述基因敲除片段电转入含pKD46质粒的大肠杆菌E.coliTHRD感受态细胞中获得阳性转化子,消除掉阳性菌中的氯霉素抗性基因后即可获得ilvI基因敲除菌;
二、ilvA基因的定点突变和过表达
<1>采用PCR技术以大肠杆菌E.coli THRD(保藏号CGMCC No.11074)基因组为模板,根据E.col iMG1655中ilvA(GeneID:948287)基因的5’端到突变处设计引物ilvA-1、ilvA-2,突变处到3’端设计引物ilvA-3、ilvA-4,扩增获取ilvA基因突变处前后的两段片段A1和A2;
<2>通过引物ilvA-2和ilvA-3完全反向互补,扩增片段重叠后将碱基序列GCTGCTGCGCTTCCT突变为GTCTGCTGCGCTTCGC,即以<1>中获得的两条扩增片段为模板通过重叠PCR获得突变后的ilvA基因;
<3>将点突变后的ilvA基因及温控表达载体PBV220(也同样可用于PBV221-ilvA或PBV222-ilvA)分别用pstI和BamHI进行双酶切后连接,连接产物转化至上述ilvI基因敲除菌中,菌落PCR鉴定后获得的阳性转化子即为本发明所述的产生α-酮基丁酸的基因工程菌;
所述PBV221-ilvA,PBV222-ilvA和PBV220-ilvA构建方法相同。
本发明的有益效果是:
上述α-酮基丁酸的发酵生产工艺,利用热诱导型表达载体过表达定点突变的L-苏氨酸脱水酶基因ilvA并敲除乙酰羟基酸合成酶III的大亚基编码基因ilvI,通过温度变化人为调控表达,操作简便、成本低,在发酵过程中通过温度控制将菌体的快速生长期和α-酮基丁酸合成期分开,使这两个阶段互不影响,从而解决发酵过程中高浓度的α-酮基丁酸(≧8g/L)严重抑制菌体生长和产酸能力等问题,达到提高菌体生物量,增加α-酮基丁酸发酵水平的目的。使用特定的产生α-酮基丁酸的基因工程菌,通过温控诱导的发酵方式,可以更加有效的解决发酵过程中α-酮基丁酸对菌体生长和产酸的抑制问题。
附图说明
图1为背景技术中α-酮基丁酸浓度梯度下基因工程菌干重曲线图谱。
图2为E.coli THRD△ilvIH的构建过程图。
图3-a为ilvI敲除重叠PCR图谱:其中M为Marker;1为ilvI基因上游498bp;3为氯霉素抗性基因片段1090bp;4为ilvI基因下游497bp;
图3-b为ilvI敲除鉴定PCR图谱:其中M为Marker;1为E.coli THRD菌株1175bp;2为消除氯霉素抗性的ilvI敲除菌1545bp;3为成功消除氯霉素抗性的ilvI敲除菌500bp。
图4为重组质粒PBV220-ilvA菌株构建过程图。
图5为重组质粒PBV220-ilvA酶切鉴定图谱:其中M为Marker;1为BαmHI和PstI双酶切PBV220空质粒3666bp;2为BαmHI和PstI双酶切重组质粒PBV220-ilvA3666bp和1561bp。
图6为α-酮基丁酸标准品(浓度10g/L)液相检测峰图。
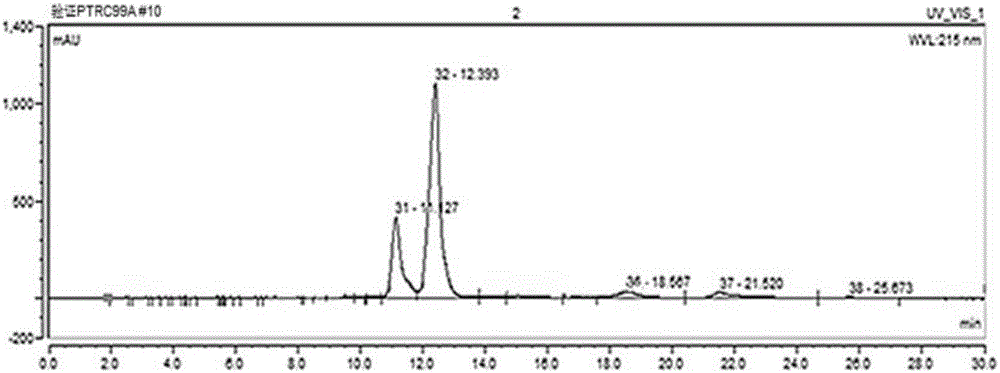
图7为摇瓶发酵24h发酵液样品液相检测峰图。
图8为7.5L罐发酵28h发酵液样品液相检测峰图。
图9为7.5L发酵罐上α-酮基丁酸积累量的发酵过程曲线。
保藏信息
分类名词:大肠埃希氏菌Escherichia coli
保藏单位名称:中国微生物菌种保藏管理委员会普通微生物中心
保藏单位地址:北京市朝阳区北辰西路1号院3号
保藏日期:2015年7月14日
保藏号:CGMCC No.11074
具体实施方式
为了使本领域的技术人员更好的理解本发明的技术方案,下面结合具体实施方式对本发明所述技术方案作进一步的详细说明。
实施例1:产生ɑ-酮基丁酸的基因工程菌株E.coli THRD△ilvIH+PBV220-ilvA的构建
1.乙酰羟基酸合成酶III大亚基编码基因ilvI的敲除
(1)根据NCBI数据库中E.col iMG1655中ilvI基因5’和3’端500bp序列设计上下游同源臂扩增引物ilvI-1、ilvI-2和ilvI-5及ilvI-6,并以E.coliTHRD菌株(保藏号CGMCC No.11074)基因组DNA为模板扩增同源臂片段。PCR扩增条件为95℃5min1个循环,94℃30s、56℃30s、72℃1min25个循环,72℃10min1个循环,反应体系为50μL。
(2)根据质粒pKD3中氯霉素抗性基因序列设计出扩增引物CM-3、CM-4,并以质粒pKD3为模板扩增氯霉素抗性基因片段,PCR扩增条件为95℃5min1个循环,94℃30s、56℃30s、72℃1.5min25个循环,72℃10min1个循环,反应体系为50μL。将(1)和(2)中PCR产物经1.0%琼脂糖凝胶电泳后切胶回收,将回收的片段分别命名为I1、CM2和I3。
(3)按1:2:1的摩尔浓度比混合I1、CM2和I3作为模板,利用引物ilvI-1、ilvI-6进行三段重叠PCR,PCR条件为95℃5min1个循环,94℃30s、56℃30s、72℃2.5min25个循环,72℃10min1个循环,反应体系为50μL。将重叠PCR产物经1.0%琼脂糖凝胶电泳后切胶回收,获得ilvI敲除片段,命名为△ilvIH。
(4)将片段△ilvIH电转化至含pKD46质粒的E.coli THRD菌株(保藏号CGMCC No.11074)感受态细胞中(电转化电压为1800V,时间为5.5ms)。迅速加入1mL SOC培养基于37℃、200rpm复苏1h后涂布于含氯霉素(25μg/mL)LB固体培养基平板上。倒置培养24h后采用ilvI敲除鉴定引物△IH-F和△IH-R通过菌落PCR鉴定阳性转化子,三段重叠片段成功整合到E.coli THRD基因组上的菌落其扩增片段约1454bp。将温敏质粒pCP20转化至上述阳性转化子以去除氯霉素抗性基因,经42℃过夜培养后,筛选能够在无抗性平板上生长而在含氯霉素平板上不生长的单菌落并采用ilvI敲除鉴定△IH-F和△IH-R进行验证,ilvI被成功敲除且丢失氯霉素抗性基因的菌株扩增片段约为500bp,获得E.coli THRD△ilvIH菌株。
构建过程如图2,图3-a,图3-b和图4为PCR验证结果,引物序列见表1。
表1ilvI基因敲除引物
2.ilvA的定点突变与过表达
(1)根据NCBI数据库中E.col iMG1655中ilvA基因序列设计ilvA-1、ilvA-2和ilvA-3、ilvA-4两对引物,以E.coliTHRD基因组DNA为模板分别扩增A1和A2两片段,PCR扩增条件为95℃5min1个循环,94℃30s、56℃30s、72℃1.5min25个循环,72℃10min1个循环,反应体系为50μL,将回收的片段分别命名为A1和A2。
(2)按1:1的摩尔浓度比混合A1和A2作为模板,利用引物ilvA-1、ilvA-4进行重叠PCR,PCR条件为95℃5min1个循环,94℃30s、56℃30s、72℃2min25个循环,72℃10min1个循环,反应体系为50μL。将重叠PCR产物经1.0%琼脂糖凝胶电泳后切胶回收,获得点突变的ilvA基因片段。
(3)将点突变的ilvA基因片段和质粒PBV220分别用PstI和BαmHI进行双酶切后连接,连接产物化转入大肠杆菌DH5α感受态细胞,挑取能在氨苄青霉素平板(100μg/mL)上生长的转化子并经鉴定引物PBV-jd1和PBV-jd2进行菌落PCR鉴定。提取阳性转化子质粒进行测序,结果与实验预期一致,表明重组质PBV220-ilvA粒构建成功。构建过程如图4,双酶切电泳图谱如图5。
(4)将重组质粒PBV220-ilvA电转入实施例1获得的ilvIH基因单敲除菌株,挑取能在氨苄青霉素平板(100μg/mL)上生长的转化子并经鉴定引物PBV-jd1和PBV-jd2进行菌落PCR鉴定,获得的阳性转化子即为本发明所述基因工程菌,命名为E.coli THRD△ilvIH+PBV220-ilvA。实例2所用引物序列见表2。
表2ilvA基因过表达引物
实施例2:产生α-酮基丁酸的基因工程菌的摇瓶发酵
<1>菌株:E.coli THRD△ilvIH+PBV220-ilvA抗性:AmpR
<2>种子培养:菌种经AmpR斜面活化两代后,无菌条件下用接种针刮1环湿菌体接于30mL含有100μg/mLAmpR的种子培养基中。37℃,200rpm培养8h,期间根据指示剂颜色变化用氨水调pH≈7.0。
<3>发酵条件:种子液培养8h后,种子液OD600=8,用灭菌的5mL移液管无菌条件下吸取3mL种子液接于27mL含有100μg/mLAmpR的发酵培养基中,接种量为10%。35℃,200rpm开始进行发酵培养,当发酵液OD600=15时,直接提高温度至42℃进行诱导表达,至发酵24h结束,期间根据指示剂颜色变化,用氨水调节pH≈7.0。
<4>利用高效液相色谱测定发酵液中α-酮基丁酸的含量,具体检测方法为:发酵液样品经13000转离心2min,上清液经0.22μm膜过滤后用UltiMate3000(Therm Scientific)高效液相测试α-酮丁酸产量,检测条件各参数为:色谱柱AminexR HPX-87H(300mm×7.8mm),流动相为5mmol/L H2SO4,流速0.5mL/min,柱温30℃,检测波长215nm,进样量为20μL。经检测,如图6和图7所示,α-酮基丁酸摇瓶产量为12.5g/L。
其中,种子培养基(g/L):葡萄糖25,酵母粉10,蛋白胨6,KH2PO41.2,MgSO4·7H2O 0.5,FeSO4·7H2O 10mg/L,MnSO4·H2O 10mg/L,VB11.3mg/L,VH 0.3mg/L,pH 6.7-7.0,115℃灭菌15min。
发酵培养基(g/L):葡萄糖30,酵母粉2,蛋白胨4,柠檬酸钠1,KH2PO42,MgSO4·7H2O 0.7,FeSO4·7H2O 100mg/L,MnSO4·H2O 100mg/L,VB11.3mg/L,VH 0.3mg/L,pH=6.7-7.0,消泡剂若干滴,121℃灭菌20min。
实施例3:生产α-酮基丁酸基因工程菌的7.5L发酵罐发酵
<1>菌株:E.coli THRD△ilvIH+PBV220-ilvA抗性:AmpR
<2>种子培养:菌种经AmpR斜面活化两代后,无菌条件下用接种针将4支斜面上的湿菌体全部接种到装有2L种子培养基的5L种子罐中,流加25%(W/V)的氨水调节种子液pH=7.0,通过提高转速和通风量使溶氧维持在30-50%左右,37℃恒温培养。
<3>发酵条件:待种子液OD600=15时,将500mL种子液转移到含有无菌发酵培养基的7.5L发酵罐中,总装液量为4L,接种量为12%。35℃培养,期间流加25%(W/V)的氨水调节发酵液pH≈6.7-7.0,提高转速和通风量使溶氧维持在30-50%左右。待发酵液OD600=25时,采用35℃→37℃→39℃→40℃(注意0.5h内完成)的升温模式进行诱导表达,之后40℃恒温发酵培养至28h发酵结束。
其中种子培养基和发酵培养基配方同实施例2中所述。
<4>利用高效液相色谱测定发酵液中α-酮基丁酸的含量,具体检测方法同实施例2中所述。经检测,如图8和图9所示,α-酮基丁酸的上罐产量为22.8g/L。
上述参照具体实施方式对该一种α-酮基丁酸的发酵生产工艺进行的详细描述,是说明性的而不是限定性的,可按照所限定范围列举出若干个实施例,因此在不脱离本发明总体构思下的变化和修改,应属本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!