1-羟基-2-(氧乙酰基十肽苯基)-四甲基咪唑啉,其合成,活性及应用的制作方法
本发明涉及1-羟基-2-[4’-氧乙酰基-Lys(Gly-Arg-Pro-Ala-Lys)-Arg-Gly-Asp-Val]苯基-4,4,5,5-四甲基咪唑啉,涉及它的制备方法,涉及它的抗血栓活性,涉及它的溶血栓活性,涉及它治疗脑缺血大鼠的作用,因而本发明涉及它在制备抗血栓药物,溶血栓药物和治疗缺血性中风药物中的应用。
背景技术:
缺血性中风是一类较常见且危害严重的脑血管疾病,特点是发病率高、病死率高、致残率高和复发率高。目前临床治疗缺血性中风面临没有有效药物的现实,尤其中风面4h以上的患者非死即残。发明对中风面4h以上的患者有效的药物是临床的重要需求。发明人曾经公开式II的咪唑啉化合物在中风面24h的大鼠缺血性中风模型上,显示优秀疗效。即连续静脉注射6天式II的咪唑啉化合物,每天1次,首次剂量为5μmol/kg,后5次的剂量为2μmol/kg具有优秀疗效。式中aa1和aa2可为同时存在,aa1存在但aa2不存在,或同时不存在;当aa1和aa2同时存在时,aa1为R(Arg),且aa2为G(Gly),A(Ala)或Q(Gln);当aa1存在但aa2不存在时,aa1为R(Arg);aa3可为S(Ser),V(Val)或F(Phe)。由于式II的咪唑啉是NO自由基,所以稳定性比较差需要改进。
发明人经过3年实验研究,发现用1羟基咪唑啉代替上式中的1,3-二氧咪唑啉可以获得稳定性好的意想不到的技术效果。按照这个发现,发明人提出了本发明。
技术实现要素:
1.本发明的内容之一是提供下式的1-羟基-2-[4’-氧乙酰基-Lys(Gly-Arg-Pro-Ala-Lys)-Arg-Gly-Asp-Val]苯基-4,4,5,5-四甲基咪唑啉
2.本发明的内容之二是提供1-羟基-2-[4’-氧乙酰基-Lys(Gly-Arg-Pro-Ala-Lys)-Arg-Gly-Asp-Val]苯基-4,4,5,5-四甲基咪唑啉的制备方法,该方法由以下方法构成:
(1)制备2,3-二甲基-2,3-二硝基丁烷;
(2)制备2,3-二甲基-2,3-二羟胺基丁烷;
(3)制备2-(4’-羟基苯基)-1,3-二羟基-4,4,5,5-四甲基咪唑;
(4)制备2-(4’-羟基苯基)-1,3-二氧-4,4,5,5-四甲基咪唑啉;
(5)按照标准方法制备Arg(NO2)-Gly-Asp(OBzl)-Val-OBz;
(6)按照标准方法制备Gly-Arg(NO2)-Pro-Als-Lys(Z)-OBz;
(7)制备1,3-二氧-2-(4’-氧乙酸乙酯-苯基)-4,4,5,5-四甲基咪唑啉;
(8)制备1,3-二氧-2-(4’-氧乙酸-苯基)-4,4,5,5-四甲基咪唑啉;
(9)制备1,3-二氧-2-[4’-氧乙酰-Lys(Boc)-OMe-苯基]-4,4,5,5-四甲基咪唑啉;
(10)制备1,3-二氧基-2-[(4’-氧乙酰-Lys-OMe)苯基]-4,4,5,5-四甲基咪唑啉;
(11)制备1,3-二氧-2-{4’-氧乙酰基-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-OMe}苯基-4,4,5,5-四甲基咪唑啉;
(12)制备1,3-二氧-2-{4’-氧乙酰基-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}苯基-4,4,5,5-四甲基咪唑啉;
(13)制备1,3-二氧-2-{4’-氧乙酰基-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Val-OBzl}苯基-4,4,5,5-四甲基咪唑啉;
(14)制备1-羟基-2-[4’-氧乙酰基-Lys(Gly-Arg-Pro-Ala-Lys)-Arg-Gly-Asp-Val]苯基-4,4,5,5-四甲基咪唑啉。
3.本发明的内容之三是评价1-羟基-2-[4’-氧乙酰基-Lys(Gly-Arg-Pro-Ala-Lys)-Arg-Gly-Asp-Val]苯基-4,4,5,5-四甲基咪唑啉的抗血栓活性。
4.本发明的内容之四是评价1-羟基-2-[4’-氧乙酰基-Lys(Gly-Arg-Pro-Ala-Lys)-Arg-Gly-Asp-Val]苯基-4,4,5,5-四甲基咪唑啉的溶血栓活性。
5.本发明的内容之五是评价1-羟基-2-[4’-氧乙酰基-Lys(Gly-Arg-Pro-Ala-Lys)-Arg-Gly-Asp-Val]苯基-4,4,5,5-四甲基咪唑啉在脑缺血24h后治疗脑缺血的活性。
附图说明
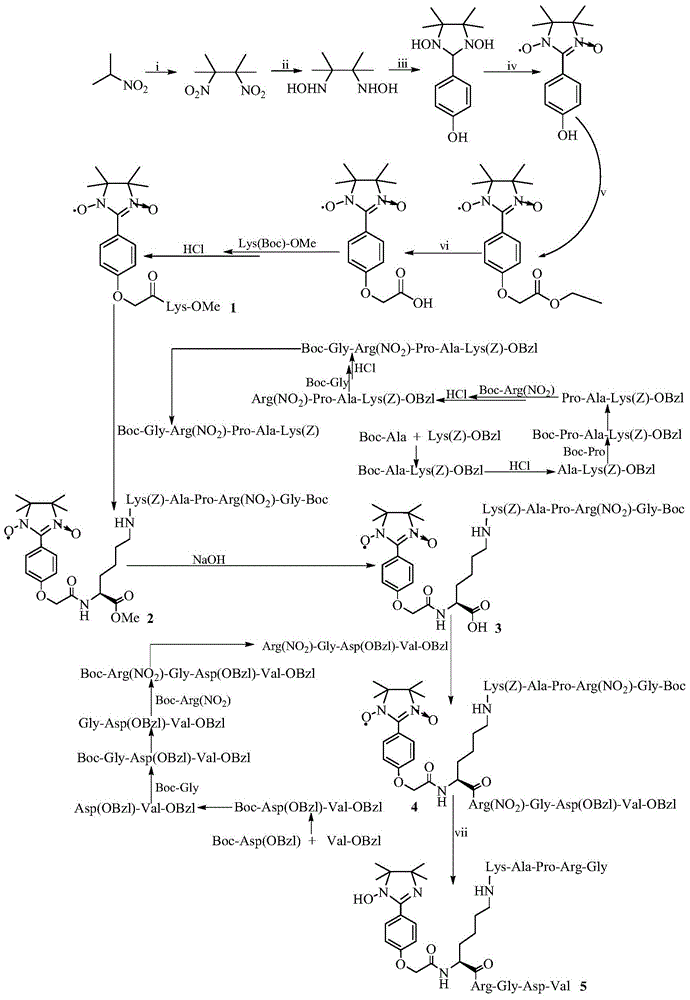
图1是1-羟基-2-[4’-氧乙酰基-Lys(Gly-Arg-Pro-Ala-Lys)-Arg-Gly-Asp-Val]苯基-4,4,5,5-四甲基咪唑啉的合成路线.i)Br2,6N NaOH;ii)Zn,NH4Cl,50%乙醇;iii)4-羟基苯甲醛,CH3OH;iv)PbO2,CH3OH;v)Br2CO2C2H5,NaH,THF;vi)2N NaOH,CH3OH;vii)TFA/TFMSA,NaNO2。
具体实施方式
为了进一步阐述本发明,下面给出一系列实施例。这些实施例完全是例证性的,它们仅用来对本发明进行具体描述,不应当理解为对本发明的限制。
实施例1制备2,3-二甲基-2,3-二硝基丁烷
69g(0.78mol)2-硝基丙烷加入130ml NaOH(6N)水溶液中,在冰盐浴搅拌下滴加20ml(0.38mol)Br2,1h内滴加完,然后加入240mL乙醇,90℃回流3h,将反应液乘热倒入800ml冰水中,滤得55g(81%)标题化合物,为无色片状结晶,Mp 110-112℃。
实施例2制备2,3-二甲基-2,3-二羟胺基丁烷
将7g(40mmol)2,3-二甲基-2,3-二硝基丁烷和4g NH4Cl混悬于80mL乙醇水溶液(50%)中,冰浴下搅拌,在3h内加入16g锌粉。锌粉加完后,撤去冰浴,继续室温搅拌反应3h,然后将反应液抽滤。滤饼用乙醇水溶液(50%)反复洗涤,合并滤液及洗涤液,用浓盐酸调节pH=2,减压蒸至泥浆状。加入适量碳酸钾,拌匀后,使用索氏提取器,氯仿为提取剂,抽提6h,提取液减压浓缩至少量,加入石油醚后析出2.60g(44%)标题化合物,为无色晶体。Mp 157-159℃。
实施例3制备1,3-二羟基-2-(4’–羟基苯基)-4,4,5,5-四甲基咪唑烷
1.22g(10mmol)对羟基苯甲醛与1.48g(10mmol)2,3-二甲基-2,3-二羟胺基丁烷溶于10mL甲醇中,室温搅拌8h后,TLC显示原料点消失。抽滤得到1.29g(51%)标题化合物,为无色晶体。EI-MS(m/z)252[M]+.1H-NMR(DMSO-d6)δ(ppm)=1.03(s,6H),1.05(s,6H),4.39(s,1H),6.70(d,J=6.9Hz,2H),7.23(d,J=6.9Hz,2H),7.63(s,1H),7.85(s,2H)。
实施例4制备1,3-二羟基-2-(4’–羟基苯基)-4,4,5,5-四甲基咪唑啉
将504mg(2mmol)1,3-二羟基-2-(4’–羟基苯基)-4,4,5,5-四甲基咪唑烷溶解于30mL甲醇中,加入3g PbO2,室温搅拌40min,TLC显示原料点消失。抽滤除去固体,滤液室温下减压蒸干,残留物柱层析纯化(氯仿洗脱),260mg(52%)标题化合物,为兰色固体。Mp 134-135℃,EI-MS(m/z)249[M]+.IR(KBr)3250,1610,1500,1490,840。
实施例5制备1,3-二氧-2-(4’-氧基乙酸乙酯-苯基)-4,4,5,5-四甲基咪唑啉
将250mg(1mmol)1,3-二羟基-2-(4’–羟基苯基)-4,4,5,5-四甲基咪唑啉、0.32mL溴代乙酸 乙酯及32mg氢化钠溶于5mL无水THF中,混合物60℃搅拌5小时,TLC显示原料点消失。室温下减压过滤,滤液减压浓缩至干,残留物柱层析(乙酸乙酯:石油醚=1:5)纯化,得到300mg(90%)目标化合物.Mp 107-109℃。
实施例6制备1,3-二氧-2-(4’-氧乙酸-苯基)-4,4,5,5-四甲基咪唑啉
向33mg(0.1mmol)1,3-二氧基-2-(4’-氧基乙酸乙酯-苯基)-4,4,5,5-四甲基咪唑啉与3mL甲醇的溶液中加7滴NaOH(2N)水溶液,室温搅拌30min,TLC显示原料点消失。将反应液减压浓缩,残留物先加2mL饱和食盐水稀释,再用2N HCl调pH 6,然后用乙酸乙酯萃取3次(3ml×3),合并的乙酸乙酯层用无水硫酸钠干燥,过滤,滤液室温下减压浓缩至干,得30mg(99%)标题化合物,为蓝色晶体。Mp 155-157℃.EI-MS(m/z)307[M]+。
实施例7制备1,3-二氧-2-[4’-氧乙酰-Lys(Boc)-OMe苯基]-4,4,5,5-四甲基咪唑啉
将307mg(1mmol)1,3-二氧-2-(4’-氧乙酸-苯基)-4,4,5,5-四甲基咪唑啉与30ml无水THF的溶液在冰浴下搅拌。向溶液加250mg(1.2mmol)DCC及135mg(1mmol)HOBt,冰浴下搅拌10min。向该溶液加由300mg(1mmol)HCl·Lys(Boc)-OMe,122mg(1mmol)N-甲基吗啉及6mL无水THF配制的溶液。得到的反应混合物室温反应24小时。TLC(乙酸乙酯:石油醚=2:1)显示HCl·Lys(Boc)-OMe消失。反应混合物减压浓缩至干,残留物用乙酸乙酯溶解,滤除不溶物。滤液依次用饱和碳酸氢钠水溶液和饱和氯化钠水溶液洗。分出的乙酸乙酯层用无水硫酸钠干燥、过滤、滤液37℃减压浓缩(这些操作在下文中统称常规处理),残留物柱层析纯化(乙酸乙酯:石油醚=2:1),得433mg(65%)标题化合物,为蓝色固体。ESI-MS(m/z)550[M+H]+。
实施例8制备1,3-二氧基-2-[(4’-氧乙酰-Lys-OMe)苯基]-4,4,5,5-四甲基咪唑啉
将625mg(1mmol)1,3-二氧-2-[4’-氧乙酰-Lys(Boc)-OMe苯基]-4,4,5,5-四甲基咪唑啉溶于15mL无水氯化氢-乙酸乙酯液(4N),室温搅拌3小时,TLC(CHCl3:MeOH,20:1)显示原料点消失。反应混合物按常规方法处理,残留物用无水乙醚析晶,得标题化合物。
实施例9制备Boc-Ala-Lys(Z)-OBzl
将473mg(2.5mmol)Boc-Ala溶于10ml无水THF。冰浴下往里加由338mg(2.5mmol)HOBt,619mg(3mmol)DCC及10mL无水THF配制的溶液。反应混合物冰浴下搅拌20分钟。向该溶液中加由936mg(2.3mmol)HCl·Lys(Z)-OBzl,232mg(2.3mmol)N-甲基吗啉先及6mL无水THF配制的溶液。得到的反应混合物室温反应24小时,TLC(CHCl3:MeOH=30:1)显示HCl·Lys(Z)-OBzl消失。反应混合物按常规处理得1.204g(97%)标题化合物,为无色固体。Mp 88-90℃.ESI-MS(m/z)565[M+Na]+。
实施例10制备HCl·Ala-Lys(Z)-OBzl:
将1.354g(2.5mmol)Boc-Ala-Lys(Z)-OBzl溶于约10ml无水氯化氢-乙酸乙酯溶液(4N),室温搅拌3小时,TLC(CHCl3:MeOH,30:1)显示原料点消失。反应混合液在室温下减压浓缩,残留物再用乙酸乙酯溶解并室温下浓缩,如此反复数次,直至除净游离的氯化氢(这些操作在下文中统称常规处理)。残留物用无水乙醚析晶,得标题化合物,直接用于下步反应。
实施例11制备Boc-Pro-Ala-Lys(Z)-OBzl
将538mg(2.5mmol)Boc-Pro溶于适量无水THF,冰浴下加入338mg(2.5mmol)HOBt,619mg(3mmol)DCC的无水THF溶液,反应20分钟。向该溶液加由1.099g(2.3mmol)HCl·Ala-Lys(Z)-OBzl,232mg(2.3mmol)N-甲基吗啉及10mL无水THF配制的溶液,室温反应24小时。TLC(CHCl3:MeOH,20:1)显示原料点消失。反应化合物按常规处理,得2.847g(98%)标题化合物.Mp 82-83℃.ESI-MS(m/z)661[M+Na] +。
实施例12制备HCl·Pro-Ala-Lys(Z)-OBzl
将1.596g(2.5mmol)Boc-Pro-Ala-Lys(Z)-OBzl溶于15mL无水氯化氢-乙酸乙酯液(4N),室温搅拌3小时,TLC(CHCl3:MeOH,20:1)显示原料点消失。反应混合物按常规方法处理,残留物用无水乙醚析晶,得标题化合物,直接用于下步反应。
实施例13制备Boc-Arg(NO2)-Pro-Ala-Lys(Z)-OBzl
冰浴下将798mg(2.5mmol)Boc-Arg(NO2),338mg(2.5mmol)HOBt,619mg(3mmol)DCC和10mL无水THF的溶液搅拌20分钟。向该溶液中加由1.322g(2.3mmol)HCl·Pro-Ala-Lys(Z)-OBzl,232mg(2.3mmol)N-甲基吗啉与5mL无水THF配制的溶液,室温反应24小时。TLC(CHCl3:MeOH,20:1)显示原料点消失。按常规处理,得1.642g(85%)标题化合物.Mp 84-85℃.ESI-MS(m/z)864[M+Na]+。
实施例14制备HCl·Arg(NO2)-Pro-Ala-Lys(Z)-OBzl
将2.099g(2.5mmol)Boc-Arg(NO2)-Pro-Ala-Lys(Z)-OBzl溶于20mL无水氯化氢-乙酸乙酯液(4N),室温搅拌3小时,TLC(CHCl3:MeOH,20:1)显示原料点消失。反应混合物按常规方法处理,残留物用无水乙醚析晶,得标题化合物,直接用于下步反应。
实施例15制备Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)-OBzl
冰浴下将438mg(2.5mmol)Boc-Gly,338mg(2.5mmol)HOBt,619mg(3mmol)DCC和10mL无水THF的溶液搅拌20分钟。向该溶液中加由1.785g(2.3mmol)HCl·Arg(NO2)-Pro-Ala-Lys(Z)-OBzl,232mg(2.3mmol)N-甲基吗啉与5mL无水THF配制的溶液, 反应24小时,得1.857g(90%)标题化合物.Mp 85-87℃.ESI-MS(m/e)920[M+Na]+。
实施例16制备Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)
将907mg(1mmol)Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)-OBzl溶于3mL甲醇中,冰浴下加入NaOH水溶液(2N),室温下搅拌30min,保持pH 12,冰浴下搅拌10min,TLC显示原料点消失。用2N HCl调pH 7,将反应液减压浓缩,残留物加2mL饱和食盐水稀释,用2N HCl调pH 2,用乙酸乙酯萃取3次(5mL×3),合并的乙酸乙酯层用无水硫酸钠干燥,室温下减压浓缩得785mg(82%)标题化合物,为无色固体.EI-MS(m/z)816[M–H]–。
实施例17制备Boc-Asp(OBzl)-Val-OBzl
冰浴下将808mg(2.5mmol)Boc-Asp(OBzl),338mg(2.5mmol)HOBt,619mg(3mmol)DCC及10ml无水THF的溶液搅拌20分钟。向该溶液加由558mg(2.3mmol)HCl·Val-OBzl,232mg(2.3mmol)N-甲基吗啉与5ml无水THF配制的溶液,室温反应24小时。TLC(CHCl3:MeOH,20:1)显示原料点消失。反应化合物按常规处理,得1.129g(96%)标题化合物,为无色油状液体。ESI-MS(m/z)512[M+H]+。
实施例18制备HCl·Asp(OBzl)-Val-OBzl
将1.278g(2.5mmol)Boc-Asp(OBzl)-Val-OBzl溶于15ml无水氯化氢-乙酸乙酯液(4N),室温搅拌3小时,TLC(CHCl3:MeOH,20:1)显示原料点消失。反应混合物按常规方法处理,残留物用无水乙醚析晶,得标题化合物,直接用于下步反应。
实施例19制备Boc-Gly-Asp(OBzl)-Val-OBzl
冰浴下将438mg(2.5mmol)Boc-Gly,338mg(2.5mmol)HOBt,619mg(3mmol)DCC和10mL无水THF的溶液搅拌20分钟。向该溶液加由1.03g(2.3mmol)HCl·Asp(OBzl)-Val-Obzl,232mg(2.3mmol)N-甲基吗啉与5mL无水THF配制的溶液,室温反应24小时。TLC(CHCl3:MeOH,20:1)显示原料点消失。反应化合物按常规处理,得1.242g(95%)标题化合物,为无色固体。Mp 66-68℃.ESI-MS(m/z)592[M+Na]+。
实施例20制备HCl·Gly-Asp(OBzl)-Val-OBzl
将1.421g(2.5mmol)Boc-Gly-Asp(OBzl)-Val-OBzl溶于15mL无水氯化氢-乙酸乙酯液(4N),室温搅拌3小时,TLC(CHCl3:MeOH,20:1)显示原料点消失。反应混合物按常规方法处理,残留物用无水乙醚析晶,得标题化合物,直接用于下步反应。
实施例21制备Boc-Arg(NO2)-Gly-Asp(OBzl)-Val-OBzl
冰浴下将798mg(2.5mmol)Boc-Arg(NO2),338mg(2.5mmol)HOBt,619mg(3mmol)DCC和10mL无水THF的溶液搅拌20分钟。向该溶液加由1.162g(2.3mmol)HCl·Gly-Asp(OBzl)-Val-Obzl,232mg(2.3mmol)N-甲基吗啉与5mL无水THF配制的溶液,室温反应24小时。TLC(CHCl3:MeOH,20:1)显示原料点消失。反应化合物按常规处理,得1.523g(86%)标题化合物,为无色固体。Mp 107-109℃.ESI-MS(m/z)793[M+Na]+。
实施例22制备HCl·Arg(NO2)-Gly-Asp(OBzl)-Val-OBzl
将1.925g(2.5mmol)Boc-Arg(NO2)-Gly-Asp(OBzl)-Val-OBzl溶于20mL无水氯化氢-乙酸乙酯液(4N),室温搅拌3小时,TLC(CHCl3:MeOH,20:1)显示原料点消失。反应混合物按常规方法处理,残留物用无水乙醚析晶,得标题化合物。
实施例23制备Boc-Asp(OBzl)-Phe-OBzl
冰浴下将808mg(2.5mmol)Boc-Asp(OBzl),338mg(2.5mmol)HOBt,619mg(3mmol)DCC和10mL无水THF的溶液搅拌20分钟。向该溶液加由668mg(2.3mmol)HCl·Phe-OBzl,232mg(2.3mmol)N-甲基吗啉与5mL无水THF配制的溶液,室温反应24小时。TLC(CHCl3:MeOH,20:1)显示原料点消失。按常规处理,得标题化合物1.222g(95%),为无色固体。Mp 79-80℃.ESI-MS(m/z)561[M+H]+。
实施例24制备HCl·Asp(OBzl)-Phe-OBzl
将1.398g(2.5mmol)Boc-Asp(OBzl)-Phe-OBzl溶于15mL无水氯化氢-乙酸乙酯溶液(4N),室温搅拌3小时,TLC(CHCl3:MeOH,20:1)显示原料点消失。反应混合物按常规方法处理,残留物用无水乙醚析晶,得标题化合物,直接用于下步反应。
实施例25制备Boc-Gly-Asp(OBzl)-Phe-OBzl
冰浴下将438mg(2.5mmol)Boc-Gly,338mg(2.5mmol)HOBt,619mg(3mmol)DCC的无水THF溶液搅拌20分钟。向该溶液加由1.141g(2.3mmol)HCl·Asp(OBzl)-Phe-OBzl,232mg(2.3mmol)N-甲基吗啉与5mL无水THF配制的溶液,室温反应24小时。TLC(CHCl3:MeOH,20:1)显示原料点消失。反应混合物按常规处理,得1.29g(91%)标题化合物,为无色固体。Mp 70-71℃.ESI-MS(m/z)640[M+Na]+。
实施例26制备HCl·Gly-Asp(OBzl)-Phe-OBzl
将1.541g(2.5mmol)Boc-Gly-Asp(OBzl)-Phe-OBzl溶于15mL无水氯化氢-乙酸乙酯液(4N),室温搅拌3小时,TLC(CHCl3:MeOH,20:1)显示原料点消失。反应混合物按常规方法处理,残留物用无水乙醚析晶,得标题化合物,直接用于下步反应。
实施例27制备Boc-Arg(NO2)-Gly-Asp(OBzl)-Phe-OBzl
冰浴下将798mg(2.5mmol)Boc-Arg(NO2),338mg(2.5mmol)HOBt,619mg(3mmol)DCC和10mL无水THF的溶液搅拌20分钟。向该溶液加由1.272g(2.3mmol)HCl·Gly-Asp(OBzl)-Phe-OBzl,232mg(2.3mmol)N-甲基吗啉与5mL无水THF配制的溶液,室温反应24小时。TLC(CHCl3:MeOH,20:1)显示原料点消失。反应混合物按常规处理,得1.637g(87%)标题化合物,为无色固体。Mp 77-79℃.ESI-MS(m/z)841[M+Na]+。
实施例28制备HCl·Arg(NO2)-Gly-Asp(OBzl)-Phe-OBzl
将2.045g(2.5mmol)Boc-Arg(NO2)-Gly-Asp(OBzl)-Phe-OBzl溶于15mL无水氯化氢-乙酸乙酯液(4N),室温搅拌3小时,TLC(CHCl3:MeOH,20:1)显示原料点消失。反应混合物按常规方法处理,残留物用无水乙醚析晶,得标题化合物。
实施例29制备1,3-二氧-2-{4’-氧乙酰基-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-OMe}苯基-4,4,5,5-四甲基咪唑啉
冰浴下将817mg(1mmol)Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z),135mg(1mmol)HOBt,250mg(1mmol)DCC和10mL无水THF的溶液搅拌20分钟。向该溶液加由480mg(1mmol)1,3-二氧-2-[(4’-氧乙酰基-Lys-OMe)苯基]-4,4,5,5-四甲基咪唑啉和100mg(1mmol)N-甲基吗啉与5mL无水THF配制的溶液,室温反应24小时。TLC(CHCl3:MeOH,40:1)显示原料点消失。反应化合物按常规处理,得680mg(52%)标题化合物,为蓝色固体。Mp 79-82℃. ESI-MS(m/z)1261[M+Na]+。IR(KBr)3319,2935,1658,1531,1448,1363,1254,1168,1053,835,749,540cm-1.
实施例30制备1,3-二氧-2-{4’-氧乙酰基-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}苯基-4,4,5,5-四甲基咪唑啉
冰浴下将1260mg(1mmol)1,3-二氧-2-{4’-氧乙酰基-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-OMe}苯基-4,4,5,5-四甲基咪唑啉溶于3mL甲醇中,加入NaOH水溶液(2N),室温下搅拌30min。保持pH 12,冰浴下搅拌10min,TLC显示原料点消失。用2N HCl调pH7,将反应液减压浓缩,残留物用2mL饱和食盐水稀释,用2N HCl调pH 2,用乙酸乙酯萃取3次(5mL×3)。合并的乙酸乙酯层用无水硫酸钠干燥,过滤,滤液室温下减压浓缩,得945mg(82%)标题化合物,为蓝色固体。EI-MS(m/z)1223[M-H]–。
实施例31制备1,3-二氧-2-{4’-氧乙酰基-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Val-OBzl}苯基-4,4,5,5-四甲基咪唑啉
冰浴下将611mg(0.5mmol)1,3-二氧-2-{4’-氧乙酰基-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys}苯基-4,4,5,5-四甲基咪唑啉,69mg(0.5mmol)HOBt,126mg(0.6mmol)DCC和20mL无水THF的溶液搅拌20分钟。向该溶液加由421mg(0.5mmol)HCl·Arg(NO2)-Gly-Asp(OBzl)-Val-OBzl,50mg(0.5mmol)N-甲基吗啉及5mL无水THF配制的溶液,室温反应24小时。TLC(CHCl3:MeOH,20:1)显示原料点消失。反应混合物按常规处理,得392mg(35%)标题化合物,为蓝色固体。Mp 147-150℃.ESI-MS(m/z)1899[M+Na]+。IR(KBr)3311,3068,2937,1661,1531,1451,1395,1254,1163,839,743,697,596cm-1。
实施例32制备1-羟基-2-[4’-氧乙酰基-Lys(Gly-Arg-Pro-Ala-Lys)-Arg-Gly-Asp-Val]苯基-4,4,5,5-四甲基咪唑啉(5)
冰浴下将396mg(0.2mmol)1,3-二氧-2-{4’-氧乙酰基-[Boc-Gly-Arg(NO2)-Pro-Ala-Lys(Z)]-Lys-Arg-(NO2)-Gly-Asp(OBzl)-Val-OBzl}苯基-4,4,5,5-四甲基咪唑啉以4mL三氟乙酸溶解。向得到的溶液中加入1mL三氟甲磺酸,冰浴条件下搅拌反应30分钟。30分钟后,将反应液转入100mL无水乙醚中,有大量沉淀析出,静置,待沉淀沉降完全后弃去上清,再向残余物中加入50mL无水乙醚,待沉淀沉降完全后再次弃去上清,减压浓缩至干,残余物用5mL蒸馏水溶解,加入150mg NaNO2,反应液颜色有蓝色逐渐变为无色(30min),将溶液的pH调至7,用Sephadex G10除盐,冻干,得到130mg(36%)标题化合物,为无色粉末。ESI-MS(m/e):1357.7497[M+H]+.1H NMR(800MHz,D2O):δ/ppm=8.687(d,J=6.4Hz,1H),8.552(d,J=6.4Hz,1H),8.493(t,J=6.4Hz,1H),8.373(d,J=5.6Hz,1H),8.28(d,J=7.2Hz,1H),8.170(d,J=7.2Hz,1H),7.998(m,2H),7.115(m,2H),4.111(m,1H),3.943(m,1H),3.900(tm,1H),3.775(s,2H),3.560(m,1H),3.113(m,7H),2.908(m,2H),2.775(dd,J=8Hz,J=5.6Hz,2H),2.223(m,1H),2.195(m,2H),1.977(m,3H),1.800(m,6H),1.710(m,3H),1.612(m,10H),1.450(m,3H),1.318(m,10H)。
实验例1评价化合物5的抗血栓活性
将雄性SD大鼠(200±20g),随机分组,每组10只,饲养1天,停止喂食过夜。灌胃给予化合物7的生理盐水溶液(剂量为1μmol/kg)或阿司匹林的生理盐水溶液(剂量为167μmol/kg)或生理盐水(剂量为10mL/kg)30min之后,大鼠用20%乌来糖的生理盐水溶液麻醉,之后手术。分离大鼠的右颈动脉和左颈静脉,将准确称重的丝线置于旁路插管,管的一端插入左静脉,另一端管插入右侧动脉并注射0.2mL肝素钠抗凝。使得血流从右侧动脉流经旁路插管进入左侧静脉,15min之后取出附有血栓的丝线称量,计算血液循环前后丝线的重量,得到的血栓重,以均值±SD mg表示并代表抗血栓活性,作t检验。数据列入 表1。结果表明口服1μmol/kg化合物5能有效地抑制血栓形成。说明本发明获得了意想不到的技术效果。
表1 1μmol/kg化合物5的抗血栓活性
n=10;a)与生理盐水相比p<0.01.
实验例2评价化合物5的溶血栓活性
SD大鼠(雄性,200±20g)按1200mg/kg的剂量腹腔注射乌拉坦生理盐水溶液进行麻醉。麻醉大鼠后将其仰卧位固定,分离其右颈总动脉,近心端处夹住动脉夹,将近心端及远心端分别穿入手术线,远心端的手术线结扎,远心端插管,将动脉夹松开,取出约1mL动脉血,置于1mL离心管中。往垂直固定的橡胶管(长15mm,内径2.5mm,外径5.0mm,管底用胶塞密封,para膜封紧)内注入0.1mL大鼠动脉血,随后在管内迅速插入一支不锈钢材质的血栓的固定螺栓(血栓固定螺旋用直径为0.2mm的不锈钢丝绕成,螺旋部分长10mm,内含15个螺圈,螺圈的直径为1.0mm托柄与螺旋相连,长约7.0mm,呈问号型)。血液凝固45min后,从玻璃管中小心取出被血栓包裹的血栓固定螺旋,精确称其重量。
旁路插管由三部分构成,中间段为长60.0mm,内径3.5mm的聚乙烯胶管;两端均为长100.0mm,内径1.0mm,外径2.0mm的相同的聚乙烯管,该管一端拉成尖管,长约10.0mm(用于插入大鼠颈动脉及静脉),外径为1.0mm,其另一端的外部套一段长为7.0mm,外径为3.5mm的聚乙烯管(用于插入中段的聚乙烯胶管内),3段管的内壁均需要硅烷化(1%的硅油乙醚溶液)。将血栓包裹的血栓固定螺旋置于中段聚乙烯胶管内,胶管的另外两端分别与两根聚乙烯的加粗端相套,保证在循环的过程中不会漏血。用注射器通过尖管端将管中注满肝素生理盐水溶液(50IU/kg),排除气泡,备用。
分离大鼠的左颈外静脉,近心端和远心端分别穿入手术线,结扎远心端的血管,在暴露的左颈外静脉上剪一小口,将上述制备好的旁路管道尖管由小口插入左颈外静脉开口处,同时远离旁路管中段(含精确称量的血栓固定螺旋)内血栓固定螺旋。用注射器通过另一端的尖管注入准确量的肝素钠的生理盐水溶液(50IU/kg),此时注射器不要撤离聚乙烯管,用动脉夹夹住注射器与聚乙烯管之间的软管。在右颈总动脉的近心端用动脉夹止血,结扎 远心端,在离动脉夹不远处将右颈总动脉剪一小口,从聚乙烯管的尖部拔出注射器,将聚乙烯管的尖部插入动脉斜口的近心端。旁路管道的两端均用4号手术缝线将动静脉固定。
用头皮针将生理盐水(3mL/kg)或尿激酶的生理盐水溶液(剂量为20000IU/kg)或化合物5的生理盐水溶液(剂量为1μmol/kg)通过旁路管的中段(含精确称量的血栓固定螺旋),扎入远离血栓固定螺旋的近静脉端,松开动脉夹,使血流通过旁路管道从动脉流向静脉。将注射器中的溶液缓慢注入血液,通过血液循环,按静脉-心脏-动脉的顺序作用于螺旋的血栓上。血液循环1h之后,从旁路管道中取出固定血栓的螺旋,精确称量。计算每只大鼠旁路管道中固定血栓的螺旋血液循环前后血栓的重量差,以均值±SD mg表示并代表溶血栓活性,作t检验。数据列入表2。结果表明1μmol/kg化合物5能有效地溶解形成的血栓。说明本发明获得了意想不到的技术效果。
表2 1μmol/kg化合物5的溶血栓活性
n=10;a)与生理盐水比p<0.01,与尿激酶相比p>0.05.
实验例3评价化合物5对缺血性中风大鼠的治疗作用
在雄性SD大鼠(体重300±20g)的颈部正中部竖直开约2cm长切口,沿胸锁乳突肌内侧缘分离出右颈总动脉,颈外动脉及颈内动脉。用无创动脉夹分别夹闭颈内动脉开口处和颈总动脉近心端,结扎颈外动脉的远心端,在颈外动脉剪1小口,松开颈总动脉近心端的动脉夹,取10μL血,之后再用无创动脉夹夹闭颈总动脉的近心端。将取得的10μL血放置在1mLEP管中常温放置30分钟使血液凝固,然后转移至-20℃冰箱中放置1小时,使血液凝块结实。大鼠用10%水合氯醛腹腔注射麻醉,剂量为400mg/kg。取出血液凝块,加入1mL生理盐水,用钢铲把血液凝块捣成大小均一的细小血栓块,制备细小血栓的悬液并转移至1mL注射器内。松开颈总动脉近心端的动脉夹,将1mL血栓混悬液缓慢从大鼠颈外动脉向近心端经过颈内动脉注入大鼠的大脑,然后结扎颈外动脉近心端,打开颈内动脉和颈总动脉处得动脉夹,恢复血流。等待苏醒。大鼠苏醒24小时后按Zealonga方法评定神经功能缺损程度。0分表示无任何神经功能缺失体征,1分表示未损伤侧前肢不能伸展,2分表示向未损伤侧行走,3分表示向未损伤侧转圈成追尾状行走,4分表示意识障碍无自主行走,5分 表示死亡。按照得分平均分组。各组大鼠经尾静脉每天注射1次化合物5剂量为1μmol/kg。连续注射6天,每天评分。结果列入表3。表3的数据表明,化合物5连续治疗6天可使10只脑缺血24小时的大鼠神经生物学评分为0分,可使2只脑缺血24小时的大鼠神经生物学评分为神经生物学评分为1分。因为不像已经公开的化合物首次剂量需要5μmol/kg,后5次维持剂量需要2μmol/kg,化合物5的6次剂量均为1μmol/kg。这样一来,首次剂量和维持剂量分别降低了5倍和2倍。
表3 化合物5连续治疗6天对脑缺血24小时大鼠神经生物学评分的影响
n=7,目标化合物剂量=1μmol/kg,鼠尾静脉注射给药。
- 还没有人留言评论。精彩留言会获得点赞!