一种制备干扰素高分子结合体IFN-POEGMA的方法与流程
本发明属于生物医药领域,具体涉及一种制备干扰素高分子结合体IFN-POEGMA的方法。
背景技术:
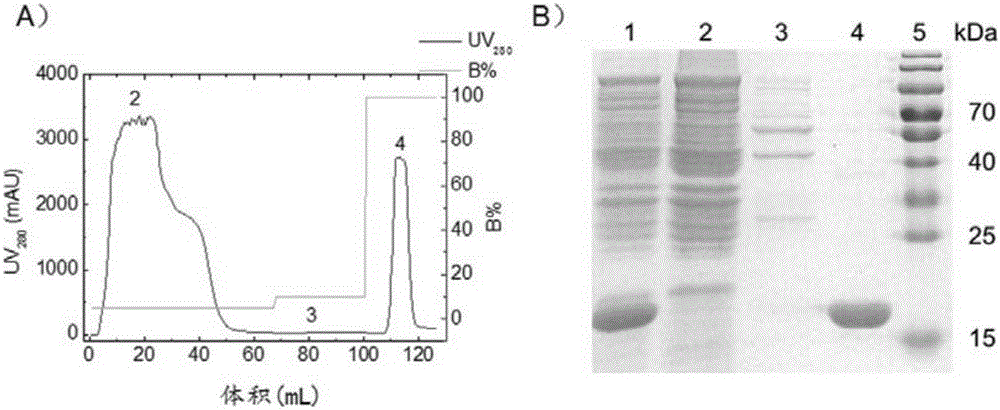
:蛋白质已经广泛应用于生物医药显影、靶向治疗以及临床诊断等多个领域。单独使用蛋白质存在半衰期短、稳定性差等问题。将蛋白质与高分子相连制备蛋白质-高分子结合体,可以有效的提高蛋白质的溶解性、稳定性、药代动力学和治疗功效并降低其免疫原性。传统的蛋白质-高分子结合体的合成方法是将预先制备好的高分子与蛋白质相连,往往存在着偶联位点不确定、效率低、产率差、产物难以分离、质量控制差、活性难以保持等诸多难题。干扰素-α2(IFN-α2)是病毒复制和肿瘤细胞生长的强效抑制剂,已被成功用于治疗病毒性肝炎和癌症等疾病。但是,IFN经系统注射给药后循环半衰期非常短,需要频繁给药且在高浓度下才能达到预期的疗效,从而导致一些毒副作用,同时给患者带来沉重的经济负担。用聚乙二醇(PEG)修饰IFN,是改善其药代动力学和提高其疗效的有效措施,被称为长效干扰素。然而,目前的PEG化干扰素存在如反应产率低、结合位点和偶联化学计量难以控制、生物活性严重降低等弊端。因此,研发反应条件温和、步骤简单、高效的位点特异性修饰方法对干扰素及其他药用蛋白质尤为重要。技术实现要素:本发明的一个目的是提供一种制备蛋白-高分子聚合物结合体的方法。本发明提供的方法,包括如下步骤:将蛋白-引发剂结合体、聚合物单体、CuCl、CuCl2、1,1,4,7,10,10-六甲基三乙烯四胺在缓冲液中进行聚合反应,得到蛋白-高分子聚合物结合体;所述蛋白-引发剂结合体为引发剂和蛋白共价连接得到的产物。上述蛋白-引发剂结合体为大分子引发剂。上述缓冲液为pH值为7.4、浓度为10mM的PBS水溶液,具体配方为:2.684gNa2HPO4·12H2O、0.34gNaH2PO4·2H2O、8.19gNaCl溶解于1L水中得到的溶液。上述方法中,所述反应体系包括:所述蛋白-引发剂结合体、所述聚合物单体、所述CuCl、所述CuCl2和所述1,1,4,7,10,10-六甲基三乙烯四胺的摩尔比为1:200-10000:10-500:10-2000:10-4000;所述蛋白-引发剂结合体、所述聚合物单体、所述CuCl、所述CuCl2和所述1,1,4,7,10,10-六甲基三乙烯四胺的摩尔比具体为1:1000-4000:25:75:125;所述蛋白-引发剂结合体中所述蛋白和所述引发剂的偶联比为1:1。在本发明的实施例中,上述蛋白-引发剂结合体为引发剂在蛋白的C端,二者通过形成酰胺键而形成结合体,具体按照包括如下步骤的方法制备:将IFN-LPETGGH6蛋白、SortaseA-H6蛋白、2-(2-(2-(2-氨基乙酰胺基)乙酰胺基)乙酰胺基)乙基2-溴-2-甲基丙酸酯、CaCl2在pH7.4浓度为50mMTris·HCl水溶液中混合,反应,得到干扰素-引发剂结合体IFN-Br;上述IFN-LPETGGH6蛋白、SortaseA-H6蛋白、上述2制备的2-(2-(2-(2-氨基乙酰胺基)乙酰胺基)乙酰胺基)乙基2-溴-2-甲基丙酸酯、CaCl2的摩尔比为2:1:50:200。上述方法中,所述聚合反应在低氧或者惰性气体氛围下进行;所述聚合反应的时间是5分钟至24小时,所述聚合反应的温度是0-80℃。上述方法中,所述引发剂连接到所述蛋白的C-端。上述方法中,所述蛋白为干扰素,所述干扰素选自干扰素α或其融合蛋白、干扰素β或其融合蛋白、干扰素γ或其融合蛋白、干扰素λ或其融合蛋白。上述方法中,所述干扰素α融合蛋白的氨基酸序列为序列表中序列2。上述方法中,所述引发剂为寡聚甘氨酸功能化的原子转移自由基聚合(ATRP)引发剂;所述寡聚甘氨酸功能化的原子转移自由基聚合(ATRP)引发剂具体为2-(2-(2-(3,4-二溴代马来酰亚胺-N-乙氧基)乙氧基)乙氧基)乙基2-溴代-2-甲基丙酸酯。上述方法中,所述聚合物单体为水溶性或生物可降解的聚合物单体;所述高分子具体为POEGMA。由上述方法制备的蛋白-高分子聚合物结合体也是本发明保护的范围。上述的蛋白-高分子聚合物结合体在制备抗肿瘤产品中的应用也是本发明保护的范围。本发明通过利用生物化学和高分子化学技术,在远离干扰素的C-末端进行修饰并通过优化过的原子转移自由基聚合(ATRP)技术原位高效生长出POEGMA(聚乙二醇甲醚甲基丙烯酸酯),产生位点特异和单修饰的干扰素-高分子结合体(即IFN-POEGMA)。这种原位ATRP技术反应条件温和、产率高、纯化步骤简单、成本低,能够有效保持生物活性并提高体外稳定性,为蛋白质修饰提供一种通用的方法。附图说明图1为干扰素-高分子结合体合成路线的示意图。图2为通过镍柱亲和层析获得IFN-LPETGGH6。图3为原位ATRP引发剂AEBM的合成及纯化过程。图4为显示了原位ATRP引发剂通过转肽酶A催化连接到IFN的C-末端示意图。图5为IFN-Br合成及纯化过程分析。图6为MALDI-TOF分析IFN-LPETGGH6和IFN-Br的分子量。图7为LC-MS/ESI分析了IFN-Br的特异性修饰情况。图8为IFN-POEGMA合成及纯化过程。图9为SDS-PAGE和GPC分析IFN-POEGMA图10为显示了通过1HNMR分析IFN-POEGMA结合体。图11为分析原位ATRP合成IFN-POEGMA的对照试验。图12为通过“graftingto”技术获得IFN-POEGMA图13为SDS-PAGE分析蛋白酶K处理IFN-POEGMA。图14为通过原位ATRP得到不同分子量的IFN-POEGMA。图15为DLS分析IFN-POEGMA和IFN-Br的水合半径。图16为CD分析IFN-POEGMA和IFN-Br的二级结构。图17为MTT法测定IFN-POEGMA、派罗欣和IFN-Br的体外生物活性。图18为IFN-POEGMA、派罗欣和IFN-Br的体外热稳定性。图19为利用DAS软件中房室消除二房模型分析IFN-POEGMA、派罗欣和IFN-Br的药物代谢动力学情况。图20为IFN-POEGMA、派罗欣和IFN-Br在肿瘤和其他组织中的分布情况。图21为IFN-POEGMA、派罗欣、IFN-Br和生理盐水抑制肿瘤生长情况。图22为裸鼠注射IFN-POEGMA、派罗欣、IFN-Br和生理盐水后的生存曲线。图23为裸鼠注射IFN-POEGMA、派罗欣、IFN-Br和生理盐水后的体重变化情况。图24为裸鼠治疗结束后肿瘤、心脏、肝脏和肾脏HE染色情况。图25为裸鼠注射IFN-POEGMA、派罗欣、IFN-Br和生理盐水后乳酸脱氢酶、肌酸激酶同工酶、谷丙转氨酶、谷草转氨酶、肌酐和尿素氮等生理指标变化情况。图26为裸鼠IFN-POEGMA、派罗欣、IFN-Br和生理盐水后红细胞、白细胞、血红蛋白和血小板等生理指标变化情况。具体实施方式下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。下述实施例中的质粒pET-25b(+)为生工生物工程(上海)股份有限公司产品。下述实施例中的TB培养基按照如下方法配置:向900mL水中加入蛋白胨12g、酵母提取物24g和甘油4mL,充分溶解后121℃高压灭菌15min,灭菌后的混合液冷却至 60℃,然后加入100mL灭菌的含170mmol/LKH2PO4和0.72mol/LK2HPO4的水溶液。下述实施例中的人Burkitt’sB淋巴瘤细胞和人卵巢癌细胞(OVCAR-3)均购自中国科学院肿瘤细胞库。下述实施例中的RMPI-1640培养基为Gibco公司产品。下述实施例中的雌性无胸腺(Nude)裸鼠为北京维通利华实验动物技术有限公司产品。雌性无胸腺(Nude)裸鼠在下文中简称裸鼠。下述实施例中的流程如图1所示。下述实施例中,利用基质辅助激光解吸电离飞行时间质谱(MALDI-TOF)、酶标仪、动态光散射(DLS)、圆二色谱(CD)等分析手段表征IFN-ELP和IFN的分子量、相变温度、水合半径和二级结构等物理化学性能。选用人Burkitt’sB淋巴瘤细胞,测试IFN-POEGMA和IFN的体外生物活性,即其体外抗肿瘤细胞增殖的能力;使用裸鼠模型,测试IFN-POEGMA和IFN在体内的药物代谢动力学,利用DAS3.0药物代谢动力学分析软件计算出药物代谢动力学参数;建立裸鼠肿瘤模型,测试IFN-POEGMA和IFN的抗肿瘤效果和药物分布。下述实施例中的定量实验,如无特殊说明均设置三次重复,结果取平均值。实施例1、制备干扰素高分子结合体IFN-POEGMA的方法一、制备干扰素-引发剂结合体1、制备IFN-LPETGGH6融合蛋白和转肽酶SortaseA-H61)重组菌的制备IFN-LPETGGH6融合蛋白的氨基酸序列为序列表中序列2,其编码基因的核苷酸序列为序列表中序列1;转肽酶SortaseA-H6的氨基酸序列为序列表中序列4,其编码基因的核苷酸序列为序列表中序列3;表达IFN-LPETGGH6融合蛋白的重组载体为将序列1所示的IFN-LPETGGH6融合蛋白编码基因插入pET-25b(+)载体的NdeI和EcoRI酶切位点中得到的载体,命名为pET-25b-IFN-LPETGGH6;表达SortaseA-H6的重组载体为将序列3所示的SortaseA-H6蛋白编码基因插入pET-25b(+)载体的NdeI和EcoRI酶切位点中得到的载体,命名为pET-25b-SortaseA-H6。表达IFN-LPETGGH6融合蛋白的重组菌为将表达IFN-LPETGGH6融合蛋白的重组载体导入E.coliBL21(DE3)pLysS(Novagen)中得到重组菌,命名为BL21/pET-25b-IFN-LPETGGH6;表达SortaseA-H6的重组菌为将表达SortaseA-H6的重组载体导入E.coliBL21 (DE3)pLysS(Novagen)中得到重组菌,命名为BL21/pET-25b-SortaseA-H6。2)IFN-LPETGGH6蛋白的表达和纯化将重组菌BL21/pET-25b-IFN-LPETGGH6在50mLTB培养基中(含100μg/mL氨苄青霉素),37℃、250rpm条件下震荡培养过夜。第二天再转接入1L新鲜TB培养基当中(盛在2L的摇瓶中,氨苄青霉素浓度为100μg/mL)进行大规模培养并诱导表达。具体步骤如下:首先在37℃,200rpm条件下震荡培养5h,随后将培养温度设为18℃,加入异丙基-β-D-硫代半乳糖苷(IPTG),终浓度为0.4mM,培养16h,收集培养液。以3000×g离心力离心收集菌体,去除上层培养液,得到重组菌BL21/pET-25b-IFN-LPETGGH6菌体。用30mL冰冷PBS重悬BL21/pET-25b-IFN-LPETGGH6菌体,再用超声仪在4℃条件下破碎细胞,然后将大肠杆菌破碎产物在4℃、14000×g离心力下离心15分钟。在收集的上清液中加入2mL聚乙烯亚胺(PEI,10%),再次离心15分钟。得到的上清液经过0.45μm滤膜过滤后,在AKTA蛋白纯化系统(AKTAPurifier10,GE)上经过镍亲和层析柱(HisTrapHP5mL)提纯,平衡缓冲液为10mMPBS、500mMNaCl,5%甘油、10mM咪唑,洗脱缓冲液为平衡缓冲液加500mM咪唑得到的混合液。收集洗脱峰对应的洗脱液,且用SDS-PAGE检测,留约21.1kDa的目标产物对应的洗脱液,为IFN-LPETGGH6蛋白。洗脱所得到的IFN-LPETGGH6蛋白经脱盐柱(HiPrep26/10Desalting)除去咪唑,同时置换到50mMTris·HCl缓冲液中。纯化样品用十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)测试纯度,并用分光光度计法(NanoDrop2000)测定了蛋白的浓度。SDS-PAGE分析样品由含有5%β-巯基乙醇的Laemmli样品缓冲液配制,浓度为1mg/mL,95℃下加热5min后,10μL样品装载到预制的10%SDS-PAGE凝胶中,垂直电泳80~100V电压下运行90min(电泳液为:25mMTris、250mMGlycine、0.1%SDS)。凝胶用考马斯蓝G-250染色处理后观察条带位置。结果如图2所示,A为通镍亲和层析纯化,UV监测线性洗脱得到目标产物IFN-LPETGGH6;B为SDS-PAGE分析IFN-LPETGGH6纯化情况;显示了IFN-LPETGGH6蛋白表达与镍亲和层析柱纯化情况。从左至右依次为上样前细胞裂解液、镍柱流穿液、50mM咪唑洗柱、500mM咪唑洗脱目的蛋白和蛋白标样,可以看出,通过大肠杆菌进行表达、镍亲和层析柱纯化获得纯度>95%的IFN-LPETGGH6蛋白,产率达到200mg/L培养菌液,可以看出,得到约21.1kDa的目标产物。3)SortaseA-H6蛋白的表达和纯化采用同样的方法处理重组菌BL21/pET-25b-SortaseA-H6,收集洗脱峰对应的洗脱液,且用SDS-PAGE检测,留约22.9kD的目标产物对应的洗脱液,为SortaseA-H6 蛋白。2、合成ATRP引发剂2-羟基乙基氨基甲酸叔丁醇酯(1.6g,10mmol),N,N-二异丙基乙基胺(1.8ml,11mmol,1.1当量)在冰水浴中溶于二氯甲烷(20ml)。在低于0摄氏度条件下滴加2-溴-2-甲基丙酰溴(1.25ml,10mmol),15分钟滴加完毕。30分钟后,移除冰水浴,并保持搅拌16小时。旋蒸移除溶剂,产物用硅胶柱纯化(二氯甲烷:乙酸乙酯=1:1)。产物为黄色油状液体,2-溴-2-甲基丙酸2-(2-(叔丁氧羰基)胺基)乙酯(C11H20BrNO4,2.13g,68.6%).1HNMR(400MHz,CDCl3):δ4.827(s,1H),4.243(m,2H),3.424(m,2H),1.945(s,6H),1.447(s,9H).ESI-massm/z:332.1([M+Na]+),334.1([M+Na]+).2-溴-2-甲基丙酸2-(2-(叔丁氧羰基)胺基)乙酯(2.13g)溶于6M-的氯化氢乙酸乙酯溶液50ml,搅拌2小时。反应结束后,过滤,产物为白色粉末,2-溴-2-甲基丙酸2-氨基乙酯盐酸盐(C6H13BrClNO2,1.69g,100%).ESI-massm/z:210.3([M–Cl+Na]+),212.3([M–Cl+Na]+).2-(2-(2-叔丁氧羰基)乙酰胺基)乙酰胺基)乙酸(289mg,1mmol),2-溴-2-甲基丙酸2-氨基乙酯盐酸盐(246mg,1mmol),EDC(288mg,1.5mmol),和N,N-二异丙基乙基胺(175μl,1mmol)溶于氯甲烷(10ml二),室温搅拌16小时。反应结束后,移除溶剂,残留白色固体一次用4毫升水洗涤2次,1毫升乙醇洗涤3次,然后用4毫升乙酸乙酯洗涤,得到白色粉末状产物,2-溴-2-甲基丙酸2-(2-(2-(2-叔丁氧羰基)乙酰胺基)乙酰胺基)乙酰胺基)乙酯(C15H26BrN3O6,269mg,56%).1HNMR(400MHz,CDCl3):δ4.261(t,2H),3.909(s,2H),3.871(s,2H),3.779(s,2H),3.544(t,2H),1.938(s,6H),1.449(s,9H).ESI-massm/z:481.2([M+H]+),483.1([M+H]+),503.2([M+Na]+),505.1([M+Na]+).2-溴-2-甲基丙酸2-(2-(2-(2-叔丁氧羰基)乙酰胺基)乙酰胺基)乙酰胺基)乙酯(108.2mg,0.225mmol)溶于6M的氯化氢乙酸乙酯溶液5ml,搅拌2小时。产物为白色粉末,2-溴-2-甲基丙酸2-(2-(2-(2-氨基乙酰胺基)乙酰胺基)乙酰胺基)乙酯盐酸盐,即ATRP引发剂AEBM(C12H22BrClN4O5,化学结构式如式1所示,84.1mg,98%).1HNMR(400MHz,D2O):δ4.196(t,2H),3.938(s,2H),3.815(s,2H),3.794(s,2H),3.456(t,2H),1.813(s,6H).ESI-massm/z:381.1([M–Cl+H]+),383.0([M–Cl+H]+)。式1如下:图3显示了原位ATRP引发剂AEBM的合成及纯化过程。最终成功获得纯度>95%的引发剂AEBM,产率为37.3%。3、通过转肽酶A酶催化获得干扰素-引发剂结合体通过转肽酶A酶催化在IFN-LPETGGH6的C-末端引入ATRP引发剂AEBM,具体如下:将上述1制备的上述IFN-LPETGGH6蛋白、SortaseA-H6蛋白、上述2制备的2-(2-(2-(2-氨基乙酰胺基)乙酰胺基)乙酰胺基)乙基2-溴-2-甲基丙酸酯、CaCl2在pH7.4浓度为50mMTris·HCl水溶液中混合,反应,得到干扰素-引发剂结合体IFN-Br;上述IFN-LPETGGH6蛋白、SortaseA-H6蛋白、上述2制备的2-(2-(2-(2-氨基乙酰胺基)乙酰胺基)乙酰胺基)乙基2-溴-2-甲基丙酸酯、CaCl2的摩尔比为2:1:50:200。具体方法如下:在10mL含有IFN-LPETGGH6(200μM)的50mMTris·HCl溶液(溶质为IFN-LPETGGH6,溶剂为pH值为7.4、浓度为50mMTris·HCl水溶液)中加入5mM2-(2-(2-(2-氨基乙酰胺基)乙酰胺基)乙酰胺基)乙基2-溴-2-甲基丙酸酯,与10mL含有100μM转肽酶A和20mMCaCl2的Tris·HCl溶液(溶质为转肽酶和CaCl2)混合,室温反应过夜,得到反应混合物。将反应混合物用阴离子交换层析(HiTrapCaptoQ5mL)纯化,得到IFN-Br;纯化采用的平衡缓冲液为20mMTris·HCl,pH7.5;洗脱缓冲液为含有1MNaCl的20mMTris·HCl水溶液,pH7.5;柱子大小为5mL,流速为2mL/min;收集洗脱峰对应的洗脱液,且用SDS-PAGE检测,留约20.5kD的目标产物对应的洗脱液,为干扰素-引发剂结合体IFN-Br。并用PBS经脱盐柱进一步去除小分子杂质。该反应效率>95%。图4显示了ATRP引发剂通过转肽酶A催化连接到IFN的C-末端,转肽酶A的半胱氨酸(Cys)作为亲核基团进行攻击,作用在IFN-LPETGGH6上识别序列LPETG的苏氨酸和甘氨酸间的肽键,使其断开(酰化作用),进而产生一个酰基酶中间物,随后三甘氨酸的氨基亲核攻击酰基酶,把AEBM共价联结到IFN-α2上。将干扰素-引发剂结合体IFN-Br用SDS-PAGE进行了验证分析,结果如图5所示,显示了IFN-Br合成及纯化过程分析,其中,A:AEX分离转肽酶A催化IFN-LPETGGH6与AEBM反应的混合物,UV监测线性洗脱得到目标产物IFN-Br;B: SDS-PAGE分析IFN-LPETGGH6与AEBM反应前后得到IFN-Br的结果。从左到右依次为SrtA-H6、IFN-LPETGGH6、反应混合物、洗脱蛋白IFN-Br、流穿样品SrtA-H6和蛋白标样。纯化产物IFN-Br的分子量用基质辅助激光解吸电离-飞行时间质谱(MALDI-TOF)测定,所用仪器为4800PlusMALDI-TOF/TOFTM分析仪(ABSCIEX)。用液相色谱-电喷雾串联质谱法(LC-MS/ESI)分析验证IFN-LPETGGH6和IFN-Br的氨基酸序列及C-末端的特异性修饰,所用仪器为Q-Exactive液质联用质谱仪(ThermoScientific)。图6为MALDI-TOF分析IFN-LPTEGGH6和IFN-Br分子量,分别为21105.7和20533.1,与理论值21105.9和20532.0符合。图7为IFN-Br的C-末端肽段EGSGGGGSLPETGGG(-Br)同位素分布情况,A图为LC-MS/ESI实验值;B图为理论预测值,理论值与实验值吻合。表1显示了LC-MS/ESI分析的IFN-Br胰蛋白酶分解后肽段氨基酸序列组成。图6、图7和表1表明利用转肽酶A能够定点定量获得干扰素-引发剂结合体IFN-Br,干扰素-引发剂结合体IFN-Br中蛋白和引发剂的偶联比为1:1。表1序列修饰电荷MH+[Da]CDLPQTHSLGSR31313.62854RTLMLLAQMR21232.69731RTLmLLAQMRM4(氧化)31248.68982TLmLLAQMRM3(氧化)21092.59123TLMLLAQMR21076.60283RISLFSCLK21066.60832ISLFSCLK2910.50798ISLFSCLKDR21181.63384DRHDFGFPQEEFGNQFQK22226.00054HDFGFPQEEFGNQFQK21954.86662AETIPVLHEMIQQIFNLFSTK22459.30058AETIPVLHEmIQQIFNLFSTKM10(氧化)32475.29734DSSAAWDETLLDK21450.66924FYTELYQQLNDLEAcVIQGVGVTETPLMKC15(脲甲基化);43359.64882FYTELYQQLNDLEAcVIQGVGVTETPLmKC15(脲甲基化);33375.64304M28(氧化)EDSILAVR2902.49407EDSILAVRK21030.58916KYFQR2741.40355ITLYLKEK21007.61278ITLYLK2750.47972KYSPCAWEVVR21337.66753YSPCAWEVVR21209.57158AEIMR1619.32322SFSLSTNLQESLR21481.76055SKEGSGGGGSLPETGGgG17(-Br)21624.64482EGSGGGGSLPETGGgG15(-Br)21409.51714二、干扰素高分子结合体IFN-POEGMA的获得1、干扰素高分子结合体IFN-POEGMA的制备在上述干扰素-引发剂结合体IFN-Br的引发剂ATRP表面合成POEGMA,方法包括如下步骤:将干扰素-引发剂结合体IFN-Br、OEGMA、CuCl、CuCl2、1,1,4,7,10,10-六甲基三乙烯四胺(HMTETA)和pH值为7.4、浓度为10mM的PBS水溶液(PBS配方为:2.684gNa2HPO4·12H2O、0.34gNaH2PO4·2H2O、8.19gNaCl溶解于1L水中),在密闭条件下反应,得到干扰素高分子结合体IFN-POEGMA;干扰素-引发剂结合体IFN-Br、OEGMA、CuCl、CuCl2、1,1,4,7,10,10-六甲基三乙烯四胺(HMTETA)的摩尔比为1:1000-4000:25:75:150;具体如下:在2.5mL含有40μMIFN-Br的PBS溶液(溶质为IFN-Br,溶剂为pH值为7.4、浓度为10mM的PBS溶液)中加入100μmol的OEGMA,通氮气15min后,加入预先溶解于1mL双蒸水中并已除空气的CuCl(2.5μmol),CuCl2(7.5μmol)和1,1,4,7,10,10-六甲基三乙烯四胺(HMTETA)(12.5μmol),密闭条件下反应1h,然后暴露在空气中终止反应,得到反应混合物。反应混合物经阴离子交换柱(HiTrapCaptoQcolumn,GEHealthcare)分离纯化出IFN-POEGMA,该纯化步骤与上述相同:纯化采用的平衡缓冲液为20mMTris·HCl,pH7.5;洗脱缓冲液为20mMTris·HCl,1MNaCl,pH7.5,柱子大小为5mL,流速为2mL/min;收集洗脱峰对应的洗脱液,且用SDS-PAGE检测,留约20 kDa的目标产物对应的洗脱液,为IFN-POEGMA1。与上述方法相同,不同是加入200μmol的OEGMA,收集洗脱峰对应的洗脱液,且用SDS-PAGE检测,留约60kDa的目标产物对应的洗脱液,为IFN-POEGMA2。与上述方法相同,不同是加入400μmol的OEGMA,收集洗脱峰对应的洗脱液,且用SDS-PAGE检测,留约120kDa的目标产物对应的洗脱液,为IFN-POEGMA3。将IFN-POEGMA1、IFN-POEGMA2、IFN-POEGMA3分别用SDS-PAGE检测,分子量分别为分子量20kDa、60kDa和120kDa,与预测大小相同。可以看出,通过改变单体OEGMA投料(100,200和400μmol)可以控制IFN-POEGMA结合体的分子量。计算产率,公式为(IFN-POEGMA结合体的质量/IFN-LPETGGH6的质量)*100%,结果如表2,IFN-POEGMA1、IFN-POEGMA2、IFN-POEGMA3的产率分别为38.4%,66.1%和78.0%。表22、干扰素高分子结合体IFN-POEGMA的结构分析图8显示了IFN-POEGMA的合成及纯化过程,其中,A为SDS-PAGE分析IFN-Br原位ATRP生长高分子POEGMA,从左往右依次为:反应混合物、纯化后的IFN-POEGMA、未反应的IFN-Br。B:AEX分离原位ATRP反应的混合物,UV监测线性洗脱得到目标产物IFN-POEGMA。结果表明阴离子交换方法能够非常有效地从ATRP反应混合物中分离IFN-POEGMA。图9为SDS-PAGE和GPC分析IFN-POEGMA,A图为SDS-PAGE分析IFN-POEGMA,从左往右依次为:纯化后的IFN-POEGMA、IFN-Br、IFN-LPETGGH6,B图为GPC分析IFN-POEGMA。图10通过1HNMR分析IFN-POEGMA结合体。为了验证POEGMA是在IFN-Br的C-末端合成的,用不含引发剂的三甘氨酸连接到IFN-α2上,然后用上述相同的条件进行了对照试验。图11显示了分析ATRP合成IFN-POEGMA的对照试验,其中,A:GPC分析IFN-Br合成IFN-POEGMA和IFN-GGG 对照试验。B:SDS-PAGE分析,从左至右依次为蛋白标样、IFN-Br进行ATRP反应、IFN-GGG对照试验和IFN-Br。对照试验采用与IFN-Br相同的反应条件对IFN-GGG用来进行ATRP反应,显示IFN-GGG并没有生长出POEGMA,说明该聚合反应仅在连接了ATRP引发剂的IFN-Br的C-末端进行,而不存在蛋白其他反应基团上的副反应。为了显示原位ATRP技术的优越性,利用“graftingto”技术合成IFN-POEGMA。Graftingto技术包含两步骤:1)合成POEGMA;2)POEGMA接枝到IFN-α2的C-末端。在上述相同条件下利用引发剂AEBM合成对应分子量的POEGMA(~60kDa),利用超滤方法除去小分子等杂志。25μMIFN-LPETGH6,12.5μM转肽酶A和500μMPOEGMA在50mMTris·HCl,150mMNaCl,10mMCaCl2,pH7.4常温过夜孵育。反应混合物经一次阳离子交换柱(平衡缓冲液为20mMTris·HCl,pH7.0;洗脱缓冲液为20mMTris·HCl,1MNaCl,pH7.0)除去转肽酶A,一次阴离子交换柱(平衡缓冲液为20mMTris·HCl,pH8.0;洗脱缓冲液为20mMTris·HCl,1MNaCl,pH8.0)除去未反应的IFN-LPETGH6和POEGMA。最后获得纯度>95%的IFN-POEGMA,产率为1.15%。图12为SDS-PAGE和GPC分析了“graftingto”技术合成IFN-POEGMA,A图为SDS-PAGE分析离子交换纯化IFN-POEGMA,从左至右分别为:蛋白标样、反应前混合物、反应24h后混合物、第一次阳离子交换柱纯化后混合物、第二次阴离子交换柱纯化后IFN-POEGMA、利用原位ATRP技术合成并纯化的IFN-POEGMA。B图为GPC技术分析“graftingto”技术。结果表明,利用“graftingto”技术合成IFN-POEGMA反应产率低,纯化困难,说明原位ATRP技术具有产率高、纯化简单、易于放大及应用等显著优势。3、干扰素高分子结合体IFN-POEGMA的物理化学表征IFN-POEGMA的分子量和多分散系数(PDI)用GPC测定,所用仪器为WatersHPLC/GPC系统连接UV检测器(Waters2489)和示差折光检测器(Waters2414)。色谱柱为AsahipakGS-520HQ和GS-320HQ或GS-520HQ串联,流动相为50mMTris·HCl缓冲液(pH7.4),检测条件为25℃,流速0.5mL/min。不同分子量的窄分布PEG标准样生成的标准曲线用来计算分子量和PDI。为了准确计算高分子POEGMA的分子量,先通过蛋白酶降解与原位高分子偶联的蛋白质再进行表征。具体步骤为:IFN-POEGMA结合体(1mg/mL)与蛋白酶K(0.5mg/mL)在50mMTris·HCl,2mMCaCl2,pH7.4,45℃孵育12h。图13显示了IFN-POEGMA可以被蛋白酶K降解IFN,再通过GPC准确表征POEGMA分子量,从左往右依次为:蛋白标样、IFN-POEGMA1、IFN-POEGMA与蛋白酶K反应前、IFN-POEGMA与蛋白酶K反应过夜后、蛋白酶K。通过已知分子量的PEG标准品被用来生成标准曲线计算IFN-POEGMA结合体的 分子量和PDI。图14显示了SDS-PAGE和GPC通过原位ATRP得到不同分子量的IFN-POEGMA。A图为SDS-PAGE分析不同分子量的IFN-POEGMA,从左往右依次是:蛋白标样、IFN-POEGMA1、IFN-POEGMA2、IFN-POEGMA3、IFN-Br。B图为GPC分析IFN-POEGMA的分子量,对应IFN-POEGMA1、IFN-POEGMA2和IFN-POEGMA3的分子量分别为23.6kDa、66.2kDa和103.7kDa,PDI分别为1.29、1.35和1.34。同时也证明可以通过调控单体/引发剂比例有效控制蛋白质-高分子结合体的分子量。在MalvernZetasizerNano-zs90上用动态光散射(DLS)方法测定IFN-POEGMA的水合半径。样品稀释在PBS缓冲液中,测试前经0.22μm孔径滤膜过滤处理。经DLS测试,IFN-Br的水合半径为2.2nm,而合成的IFN-POEGMA1、IFN-POEGMA2和IFN-POEGMA3的水合半径分别为5.9nm、10.6nm和15.2nm。图15显示了DLS分析IFN-POEGMA。用核磁共振仪(1HNMR)表征蛋白质-高分子的化学结构。IFN-POEGMA样品经冷冻干燥后,溶解于D2O,在JEOLECX-400400MHz核磁共振光谱仪上进行分析。通过1HNMR谱图证实了蛋白分子上成功合成了POEGMA。图10显示了通过1HNMR分析IFN-POEGMA2结合体。IFN-POEGMA2的二级结构用圆二色性谱分析测得。样品用水溶液稀释至0.18mg/mL(1.2μM),用Pistarπ-180(AppliedPhotophysics有限公司)在190-250nm波长范围内进行紫外扫描分析。图16显示了圆二色性色谱分析IFN-Br、IFN-POEGMA的二级结构。对IFN-Br和IFN-POEGMA结合体做了圆二色性谱分析,均在190-260nm波长范围内的圆二色谱都呈现出同样的209/219nm双峰曲线,且与IFN-Br曲线重叠良好,表明在蛋白质上生长原位高分子对蛋白质分子的二级结构没有明显影响。用二喹啉甲酸法(BCA)测定IFN-POEGMA的蛋白浓度,已知浓度的牛血清白蛋白为标准样,具体步骤按照说明书。三、对照方法制备在2.5mL含有40μMIFN-Br的PBS溶液(溶质为IFN-Br,溶剂为pH值为7.4、浓度为10mM的PBS溶液)加入一定量的OEGMA(OEGMA与IFN-Br的摩尔比分别为500:1)和11OμL维生素C水溶液(4Omg/mL),通氮气15min后,加入10μL的氯化亚铜和N,N,N`,N'`,N'`一五甲基二乙烯基三胺(PMDTA)(浓度分别为100mM和300mM)混合物储液,密闭条件下反应lh,然后暴露在空气中终止反应,得到反应混合物。反应混合物经阴离子交换柱(HiTrapCaptoQcolumn,GEHealthcare)分离纯化(纯化方法同前)出IFN-POEGMA,收集8mL柱体积的为干扰素高分子结合体IFN-POEGMA(对照组);将IFN-POEGMA(对照组)用SDS-PAGE检测和GPC测定,分子量为60.0kDa。计算产率,公式为(IFN-POEGMA结合体的质量/IFN-LPETGGH6的质量)*100%,结果如表3,IFN-POEGMA(对照组)的产率为18.9%。表3可以看出,本发明的二的方法与三的对照方法制备的同样分子量的IFN-POEGMA结合体比较,本发明的二的方法制备的IFN-POEGMA结合体的产率远远高于三的对照方法,说明本发明的方法效果好。实施例2、干扰素高分子结合体IFN-POEGMA的功能验证1、IFN-POEGMA的体外生物活性和热稳定性测试本发明中IFN-POEGMA(选择实施例2的二的方法制备的IFN-POEGMA2,即分子量为66.2kDa进行表征,下均同)的抗细胞增殖活性采用MTT方法测定。选用了人Burkitt’sB淋巴瘤细胞(DaudiB),因为该细胞对IFN-α2具有较高的敏感。DaudiB细胞在含有15%FBS、50U/mL盘尼西林和50μg/mL链霉素的RMPI-1640中培养一段时间后,在96孔板中接种一定浓度的细胞悬浮液(50μL/孔,7500个细胞),将IFN-Br、派罗欣和纯化后的IFN-POEGMA结合体样品系列稀释,各50μL加入96孔培养板中,设阴性对照(不含IFN-α2)和空白对照(只含培养液),37℃,5%CO2培养72h,加MTT溶解液20μL/孔,3h后用酶标仪测定各孔490nm波长的吸收值,比较不同样品处理后细胞增殖的程度。图17显示了MTT测定IFN-POEGMA的体外生物活性,表4显示了IFN-Br、派罗欣和IFN-POEGMA的体外生物活性,IFN-Br、派罗欣和IFN-POEGMA的IC50分别为11.6pg/mL、195.90pg/mL和27.37pg/mL,活性保持率分别为100%、5.92%和42.38%。综上可知,IFN-POEGMA体外抗肿瘤活性远高于派罗欣,表明原位ATRP没有严重降低IFN的活性,为体内抗肿瘤活性测试提供了依据。表4本发明表征了IFN-POEGMA的热稳定性。样品(200μg/mL)置于37℃1,2,4,6,8h,1,2,3,5,7,8和10天后,利用MTT法测定其抗肿瘤细胞增殖活性。如图18,IFN-POEGMA放置37℃5天活性保持在95%以上,到第10天仍然保持80%以上活性,相反,IFN-Br活性迅速下降,8h后仅保持26%的活性,24h后活性消失。该数据表明,经过POEGMA修饰后的IFN具有很好的体外热稳定性。2、IFN-POEGMA的药物代谢动力学测试以下所有动物实验均在清华大学关于动物实验的各项规定指导下完成。本发明中利用Bal/cnude裸鼠模型经尾静脉注射IFN-Br、派罗欣和IFN-POEGMA,测定了血液中干扰素浓度随时间变化的情况,并利用DAS软件进行数据分析。在药物处理期前,9只雌性裸鼠观察一段时间后随机分成3组。以1mg/kg体重剂量尾静脉注射IFN-POEGMA、派罗欣及未修饰IFN-Br对照物,然后在设定的时间点(1,5,15,30min,1,3,6,24,48,72和96h)尾静脉取血20μL(采血管预先用肝素钠(江苏万邦生化医药股份有限公司产品)浸润并烘干),室温静置1h,在4℃、3000×g下离心收集上层血浆,保存于﹣80℃低温冰箱。用人IFN-α2ELISA试剂盒(PBLinterferonsource)根据说明书测定血清中的IFN-α2含量。利用DAS3.0药物代谢动力学分析软件计算IFN-POEGMA、派罗欣和IFN-Br的药物代谢动力学参数。表5和图19分别显示了IFN-POEGMA、派罗欣和IFN-Br在裸鼠中药物代谢动力学情况。IFN分布半衰期(t1/2α)消除半衰期(t1/2β)分别为0.42h和1.58h,在给药5分钟后,血液中干扰素的浓度即迅速下降到初始剂量的50%以下,24h后残留浓度不足初始浓度的0.1%。而IFN-POEGMA在体内的浓度是逐渐减少的,其初始半衰期和消除半衰期分别为2.09h和56.34h,是IFN的35倍左右。到给药72h后,仍有20%以上的残留。IFN-POEGMA的药时曲线下面积(AUC0-∞)是IFN的167倍。以上结果表明,与未修饰的IFN相比,IFN-POEGMA结合体的消除半衰期和体内平均存留时间明显延长,药时曲线面积显著增加,清除率显著降低,与派罗欣具有相近的体内循环半衰期。表53、IFN-POEGMA体内组织分布情况本发明中利用移植了卵巢癌细胞的裸鼠测定了给药2h,24h和4d后肿瘤中残留的干扰素的浓度。人卵巢癌细胞(OVCAR-3)在含有10%FBS,50U/mL盘尼西林和50μg/mL链霉素的RMPI-1640培养基中培养一段时间后,用胰蛋白酶消化剥离,经PBS洗涤,重悬于不含上述添加物的RMPI-1640培养基并与BDMatrigelMatrix等体积混合物中,0.2mL单细胞悬液(5×106个细胞)接种于裸鼠左后肢股骨处背部皮下,培养30天后形成100-200mm3大小的实体瘤肿块。将裸鼠随机分成3组,IFN-POEGMA、派罗欣、IFN-Br以尾静脉注射打入裸鼠体内,给药剂量为10μg/20g体重。给药2h,24h和4d后处死裸鼠,收集心脏、肾脏、肝脏、脾脏、肺脏、胰腺、胃、肌肉、小肠和肿瘤等组织器官。用提取缓冲液 (PBS含1mMEDTA,0.5%TritonX-100,0.5%脱氧胆酸钠,1mMPMSF,按1:100比例稀释的蛋白酶抑制剂混合物和磷酸酶抑制剂混合物(Sigma-Aldrich))破碎后,离心取上清提取液。组织中IFN的浓度用ELISA方法定量测定。图20为IFN-POEGMA在各组织中的累积情况,说明IFN-POEGMA在心脏、肾脏、肝脏、脾脏、肺脏、胰腺、胃、肌肉、小肠和肿瘤中均能够有效累积。其中,A图为IFN-POEGMA、派罗欣、IFN-Br在肿瘤中累积情况,可以看出,IFN-POEGMA组肿瘤中干扰素的浓度和IFN组肿瘤中干扰素的浓度相比,注射2h为10.5倍,注射24h为133.2倍,注射4d后IFN-POEGMA仍有15%的残留。B图为IFN-POEGMA、派罗欣、IFN-Br在其他组织中累积的情况。表明,IFN-POEGMA能够有效利用增强通透和滞留效应,使干扰素在肿瘤组织中聚集、在血液循环中半衰期延长,从而提高干扰素在体内的生物利用度和抗肿瘤功效。4、裸鼠模型测试IFN-POEGMA体内抗肿瘤活性IFN-POEGMA的体内生物活性用动物移植性肿瘤实验法测定。按上述方法,OVCAR-3细胞接种于裸鼠左后肢股骨处背部皮下,培养3周天后形成实体瘤肿块(~20mm3),从而建立裸鼠肿瘤模型。将26只裸鼠分成4组,IFN-POEGMA、派罗欣及对照品(IFN-Br为阳性对照,生理盐水为阴性对照)以尾静脉注射打入裸鼠体内,给药剂量为20μg/20g体重,每周注射一次,直至生理盐水组、IFN-Br组和派罗欣组的裸鼠全部死亡。在本实验中裸鼠死亡包括自然死亡和安乐处死,安乐处死是指裸鼠肿瘤生长超过500mm3或体重下降超过15%,被注射巴比妥钠类药物处死。每周观察裸鼠生存状态以及肿瘤生长情况,动态测量裸鼠体重和肿瘤体积随时间的变化。为检测IFN-POEGMA的毒性,给药三次后,处死小鼠,并收集肿瘤、心脏、肝脏和肾脏,固定在4%甲醛溶液中,切片后通过标准方法进行HE染色,观察器官的组织学形态。结束治疗后,通过眼球取血,测量乳酸脱氢酶、肌酸激酶同工酶、谷丙转氨酶、谷草转氨酶、肌酐、尿素氮、红细胞、白细胞、血小板、血红蛋白等基本生理指标。实验结果表明(图21和图22),实验期间,生理盐水组的裸鼠的肿瘤体积快速增大,注射第38天,裸鼠肿瘤体积均超过500mm3,生存中值仅为35天;实验期间,IFN-Br组的裸鼠的肿瘤体积逐渐增大,注射第45天IFN组的裸鼠肿瘤体积也超过500mm3,生存中值为38天,没有体现明显的抗肿瘤活性;实验期间,派罗欣组的裸鼠的肿瘤体积逐渐增大,注射第56天IFN组的裸鼠肿瘤体积也超过500mm3,生存中值为50.5天,有一定的抗肿瘤活性;实验期间,IFN-POEGMA组的裸鼠的肿瘤体积基本没有变化,同时,有75%的小鼠肿瘤消失,只有2只小鼠肿瘤缓慢增大,直到77天和88天肿瘤体积才超过500mm3。以上数据表明,IFN-POEGMA能够有效地抑制肿瘤的生长,具有非常 好的体内抗肿瘤活性,甚至优于目前市售药品派罗欣。实验结果还表明,实验期间,IFN-POEGMA组的裸鼠体重略有增加(图23),表明IFN-POEGMA对裸鼠没有明显的副作用。图24为给药3次后小鼠的肿瘤、心脏、肝脏、肾脏的组织学切片,可以看出,注射IFN-POEGMA后,小鼠肿瘤细胞间隙出现空穴,胞浆和细胞核形态不明显,细胞坏死,大块细胞膜脱落,与对照组差异明显。同时,心脏、肝脏和肾脏的细胞形态完整,无明显细胞坏死,与对照组组织学形态无明显差异。图25和图26分别为结束治疗后小鼠的生化和血液指标,与生理盐水组、IFN-Br组和派罗欣组的裸鼠相比,IFN-POEGMA组的裸鼠的乳酸脱氢酶、肌酸激酶同工酶、谷丙转氨酶、谷草转氨酶、肌酐、尿素氮、红细胞、白细胞、血小板、血红蛋白等基本生理指标没有显著差别。以上数据表明IFN-POEGMA不会对裸鼠体内器官造成明显毒性。本发明的方法原位ATRP合成技术产率高,纯化简单,易于工业化,制备的干扰素-POEGMA结合体具有较高的生物活性保存率,显著延长的半衰期和有效的肿瘤抑制作用,与市售药物派罗欣体内外活性相比表现出更加优异的功效。ATRP合成技术可望取代PEG化成为修饰蛋白药物以提高药物稳定性、改善药物代谢动力学和增强治疗功效的新方法。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1