用于高伯羟基多元醇的双催化剂系统的制作方法
实施例涉及使用双催化剂系统制造具有高伯羟基含量的聚醚多元醇的方法。
背景技术:
通过在起始剂化合物和催化剂存在下聚合环氧烷而制备聚醚多元醇。起始剂化合物具有一种或多种官能团,环氧烷可与所述一种或多种官能团反应以开始形成聚合物链。起始剂化合物可影响分子量并产生所得聚醚多元醇将具有的羟基数量。关于用于形成聚醚多元醇的催化剂,制造朝着使用双金属氰化物(dmc)催化剂代替碱金属催化剂(如koh基催化剂)的趋势前进。然而,如在美国专利第6,482,993号中所论述的,使用dmc催化剂的缺点为需要诱导期。不同于碱金属催化剂,dmc催化剂并不一经添加氧化物和起始剂化合物就立刻开始聚合。因此,已经提出在添加dmc催化剂之前使用金属全氟烷基磺酸酯催化剂。另外,如在国际公布第wo2012/134849号中论述的方法,已经提出将超强酸和dmc催化剂结合使用(例如,以便制备相比于常规半分批基催化的方法而具有改进的和/或缩短周期时间的短链多官能聚醚多元醇和/或制备在半分批和/或连续进料方法中并不需要结束步骤的短链多官能聚醚多元醇)。首先使用额外催化剂可干扰dmc催化剂诱导的催化反应。因此,存在对于双催化剂系统的需要,所述双催化剂系统使得发生对聚合反应的dmc催化剂诱导的催化反应而没有来自它他催化剂的干扰。
另外,存在对于双催化剂多元醇制备方法的需要,所述方法使得对于获得高伯羟基含量的聚醚多元醇(即,以羟基的总数计,伯羟基含量大于60%)的改进控制。另外,存在对于双催化剂系统的需要,所述双催化剂系统除了高伯羟基含量之外,还可提供对聚合反应的精密控制以便使用较低分子量起始剂化合物(例如,在高进料速度下提供以便实现反应时间的整体改进)得到高数均分子量(即,大于2500g/mol的mn)的聚醚多元醇。
技术实现要素:
可通过提供制备高伯羟基含量和高数均分子量多元醇的方法来实现实施例,所述方法包括制备包括第一催化剂和数均分子量小于1,000g/mol的低分子量聚醚多元醇的混合物,所述聚醚多元醇衍生自环氧丙烷、环氧乙烷或环氧丁烷,并且第一催化剂为双金属氰化物催化剂;将混合物设定成具有第一温度,在第一温度下将选自环氧丙烷、环氧乙烷和环氧丁烷中的至少一种添加到混合物,并且使混合物反应以形成反应混合物;将第二催化剂添加到反应混合物,所述第二催化剂为具有通式m(r1)1(r2)1(r3)1(r4)0或1的路易斯酸(lewisacid)催化剂,而m为硼、铝、铟、铋或铒,r1和r2各自独立地包括氟取代的苯基或甲基,r3包括氟取代的苯基或甲基或官能团或官能聚合物基团,任选的r4为官能团或官能聚合物基团;和将包括第二催化剂的反应混合物设定成具有小于第一温度的第二温度并且在第二温度下添加额外的选自环氧丙烷、环氧乙烷和环氧丁烷的至少一种到反应混合物,使得形成具有至少60%的伯羟基含量和大于2,500g/mol数均分子量的所得多元醇。
附图说明
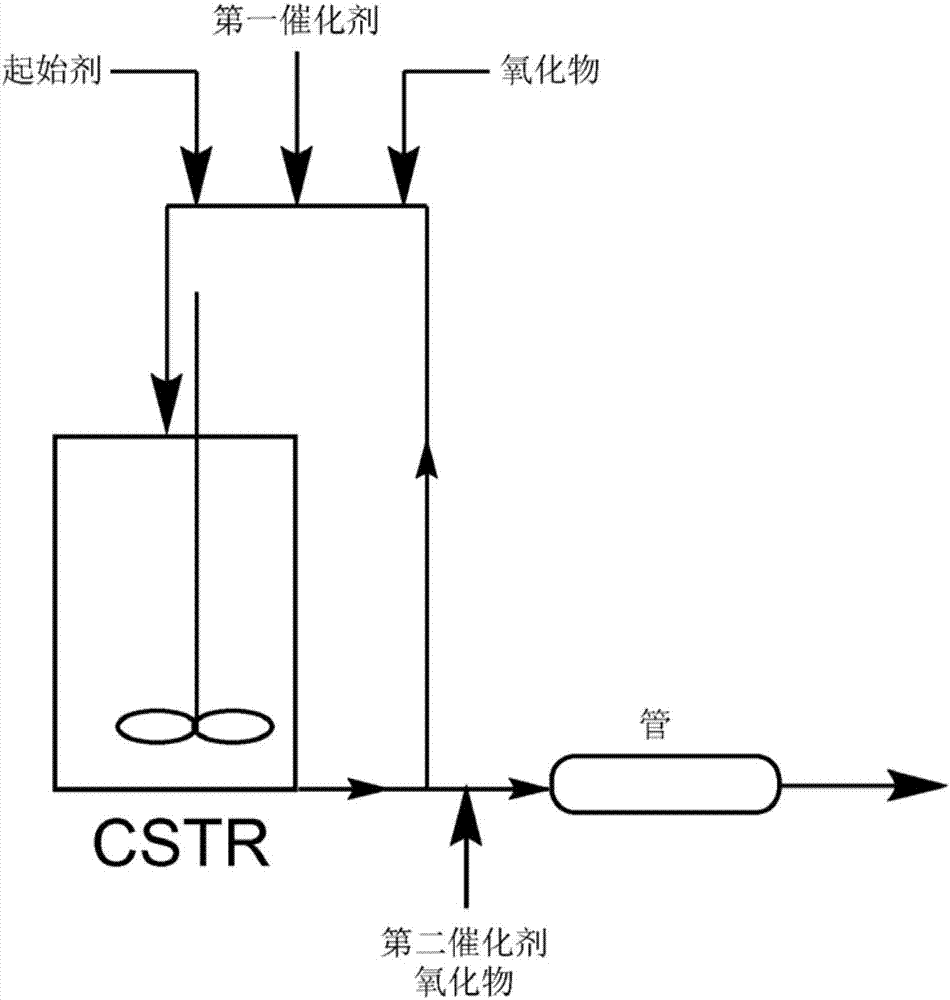
图1示出用于制备根据实施例的多元醇的例示性半分批方法示意图。
图2示出用于制备根据实施例的多元醇的例示性连续方法示意图。
具体实施方式
双金属氰化物(dmc)催化剂被视为高活性烷氧基化催化剂,其可在极低浓度(例如,以用于形成丙氧基化聚醚多元醇的组合物的总重量计,小于50ppm)下使用以实现氧化物如环氧丙烷、环氧乙烷和环氧丁烷的快速烷氧基化。举例来说,如美国专利第6,531,566号的实例1中所示,使用氢氧化钠基碱金属催化剂导致十二小时的反应时间。然而,十二小时反应时间在制造方法设定中可被视为不利的,而通过使用dmc催化剂可减小总反应时间。如在国际公布第wo2012/091968号中所论述的,某些基本上不需要活化时间的路易斯酸已经评估为聚合催化剂。然而,路易斯酸可快速变得失活而可能不能够制备高分子量聚合物或获得环氧烷到聚合物的高转化率。酸基催化剂如氢氧化钠和路易斯酸的另一缺点在于,可需要处理如过滤和/或酸修整/中和(例如,如在美国专利第5,468,839号中所论述的)以减少所得产物的酸含量。然而,使用dmc催化剂和/或足够低量的酸基催化剂(如路易斯酸)可消除对于此类处理的需要。在例示性实施例中,用于形成聚醚多元醇(如环氧丙烷类多元醇)的双催化剂系统使用以少量的dmc催化剂和硼基路易斯酸催化剂这两者,使得对于所得聚醚多元醇不需要过滤和酸修整/中和。
使用环氧烷(例如,使用环氧丙烷和排除使用任何环氧乙烷和环氧丁烷)进行dmc催化的丙基化反应的典型条件可产生具有高程度仲羟基(例如,大于90%)的多元醇。所述结果为使用dmc催化剂的特性。另外,如在国际公布第wo2012/09196号中所论述的,通过路易斯酸催化制备的聚(环氧丙烷)聚合物倾向于具有约50%仲羟基和50%伯羟基。然而,寻求较高的伯羟基含量(即,至少60%)。伯羟基意指(位于末端的含羟基基团(例如,在聚环氧烷多元醇如聚环氧丙烷多元醇上),而仲羟基意指位于非末端的含羟基基团(例如,在聚环氧烷多元醇如聚环氧丙烷多元醇上)。
实施例涉及使用双催化剂系统形成聚醚多元醇(例如,环氧丙烷基多元醇),所述双催化剂系统组合了使用高活性dmc催化剂的优势,获得高伯羟基含量(即,大于60%)和相对高数均分子量(即,大于2,500g/mol如2,600g/mol到12,000g/mol,3,000g/mol到6,000g/mol等)。
双催化剂系统例如以下面的方式利用dmc催化剂和路易斯酸催化剂:
使用双催化剂系统的方法包括初始地添加dmc催化剂和随后添加路易斯酸催化剂,所述路易斯酸催化剂单独提供且允许在比添加dmc催化剂的温度更低的温度下反应。
dmc催化剂
例示性双金属氰化物催化剂在例如,国际公布第wo2012/09196号中有所论述。所述dmc催化剂例如本领域中已知的那些可用于双催化剂系统。具体来说,dmc催化剂为作为双催化剂系统的一部分提供的第一催化剂,在所述双催化剂系统中,提供至少第一催化剂和在第一催化剂之后的第二催化剂。
举例来说,dmc催化剂可由式1表示:
mb[m1(cn)r(x)t]c[m2(x)6]d·nm3xay(式1)
其中m和m3各自为金属;m1为与m不同的过渡金属,每一个x表示除氰离子之外的与m1离子配位的基团;m2为过渡金属;a表示阴离子;b、c和d是反映静电中性络合物的数值;r为4到6;t为0到2;x和y为使金属盐m3xay中电荷平衡的整数,并且n为零或正整数。前述式不反映中性络合剂的存在,如常常存在于dmc催化剂络合物中的叔丁醇。m和m3各自为独立地选自zn+2、fe+2、co+2、ni+2、mo+4、mo+6、al+3、v+4、v+5、sr+2、w+4、w+6、mn+2、sn+2、sn+4、pb+2、cu+2、la+3和cr+3的群组的金属离子,其中zn+2为优选的。m1和m2各自独立地选自fe+3、fe+2、co+3、co+2、cr+2、cr+3、mn+2、mn+3、ir+3、ni+2、rh+3、ru+2、v+4、v+5、ni2+、pd2+、和pt2+的群组。根据示例性实施例,在加三氧化态中的那些较多用作m1和m2金属。举例来说,可使用co+3和/或fe+3。
例示性阴离子a包括(但不限于)卤离子,如氯离子、溴离子和碘离子;硝酸根;硫酸根;碳酸根;氰离子;乙二酸根;硫氰酸根;异氰酸根;过氯酸根;异硫氰酸根;烷磺酸根,如甲磺酸根;芳基磺酸根,如对甲苯磺酸根;三氟甲烷磺酸根(三氟甲磺酸根)以及c1-4羧酸根。举例来说,可使用氯离子。r为4、5或6(例如,4或6,或6);t为0或1。在例示性实施例中,r+t将等于6。
在例示性实施例中,dmc催化剂为六氰钴酸锌催化剂络合物。dmc催化剂可与叔丁醇络合。用于例示性实施例的dmc催化剂可为包括一种或多种dmc催化剂的混合催化剂。混合催化剂可任选地包括非dmc催化剂,其中所述dmc催化剂占混合催化剂总重量的至少75wt%。混合催化剂可排除稍后添加在催化剂系统中的路易斯酸催化剂中的任一种。
路易斯酸催化剂
金属类路易斯酸催化剂具有通式m(r1)1(r2)1(r3)1(r4)0或1,而m为硼、铝、铟、铋或铒,r1和r2各自独立地包括氟取代的苯基或甲基,r3包括氟取代的苯基或甲基或官能团或官能聚合物基团,任选的r4为官能团或官能聚合物基团。氟取代的苯基意指包括用氟原子替换的至少一个氢原子的苯基。氟取代的甲基意指包括用氟原子替换的至少一个氢原子的甲基。r1、r2和r3可包括氟取代的苯基或可主要由氟取代的苯基组成。r1、r2和r3可包括氟取代的甲基,例如,以与硫氧化物(例如,三氧化硫)键结的氟取代的甲基的形式。在通式中m可作为金属盐离子或作为化学式的一体地键结的一部分而存在。
官能团或官能聚合物基团可为路易斯碱,其与路易斯酸催化剂(例如,硼基路易斯酸催化剂或三氟甲磺酸金属盐催化剂)形成络合物。官能团或官能聚合物基团意指包含以下中的至少一种的分子:醇、烷芳基、具有1至12个碳原子的直链或支链烷基、环烷基、丙基、丙基氧化物、硫醇、有机硅烷、有机硅氧烷、肟、能够充当与另一个硼原子的共价桥键的亚烷基、能够充当与另一个硼原子的共价桥键的二价有机硅氧烷基团和其经取代的类似物。举例来说,官能团或官能聚合物基团可具有式(oyh)n,而o为o氧,h是氢,而y为h或烷基。然而,可使用可与路易斯酸催化剂如硼基路易斯酸催化剂或三氟甲磺酸金属盐组合的其它已知官能聚合物基团。
根据例示性实施例,路易斯酸催化剂为硼基路易斯酸催化剂,其具有通式b(r1)1(r2)1(r3)1(r4)0或1,而r1和r2各独立地为氟取代的苯基,r3为氟取代的苯基或官能团或官能聚合物基团,任选地r4为官能团或官能聚合物基团。
在例示性实施例中,硼基路易斯酸为三(五氟苯基)硼烷。
例示性三(五氟苯基)硼烷络合物具有以下结构,而ipro为异丙氧基。
路易斯酸催化剂可为三氟甲磺酸金属盐。举例来说,三氟甲磺酸金属盐具有通用m(r1)1(r2)1(r3)1(r4)0或1,而m为铝、铟、铋或铒,而r1、r2和r3各自为cf3so3。路易斯酸催化剂可在较低温度范围(例如,60℃-110℃)下具有活性。例示性参考文献包括美国专利第4687755号;williams,d.bg.;lawton,m.三氟甲磺酸铝:一种用于通过醇对环氧化物的开环的显著路易斯酸催化剂(aremarkablelewisacidcatalystfortheringopeningofepoxidesbyalcohols).有机生物分子化学(org.biomol.chem).2005,3,3269-3272;khodaei,m.m.;khosropour,ar.;ghozati,k.四面体学报(tetrahedronlett)2004,45,3525-3529;dalpozzo,r.;nardi,m.;oliverio,m.;paonessa,r.;procopio,a.三氟甲磺酸铒(iii)为用于通过对环氧化物的开环合成β-烷氧基醇、1,2-二醇和β-羟基硫化物的高效催化剂(erbium(iii)triflateisahighlyefficientcatalystforthesynthesisof).合成()2009,3433-3438。
用于例示性实施例的路易斯酸催化剂可为混合催化剂,其包括一种或多种路易斯酸催化剂(例如,各自具有通式b(r1)1(r2)1(r3)1(r4)0或1,而r1和r2各独立地为氟取代的苯基或甲基,r3为氟取代的苯基或甲基或官能团或官能聚合物基团,任选的r4为官能团或官能聚合物基团)。混合催化剂可任选包括其它催化剂,其中具有通式的路易斯酸催化剂占混合催化剂总重量的至少75wt%。所添加的混合催化剂可排除任何dmc基催化剂。在低温下具有活性的例示性其它金属基路易斯酸可包括为双催化剂系统和/或混合催化剂的一部分。例示性金属基路易斯酸是基于铝、硼、铜、铁、硅、锡、钛、锌和锆中的一个。
使用双催化剂系统
在实施例中,低羟基当量起始剂化合物(如具有小于1000g/mol的数均分子量的全部环氧丙烷多元醇)的烷氧基化不能从起始剂化合物直接进行到成品聚醚多元醇。举例来说,由于在聚合早期阶段期间高浓度严的羟基和起始剂化合物严重地抑制初始催化剂活性,这可导致催化剂诱导的失败或催化剂在烷氧基化方法早期过早失活。减少这些情况发生涉及催化剂在第一温度下的活性和通过在30分钟或更少的时段内将环氧丙烷、环氧乙烷和环氧丁烷中的至少一种缓慢添加到包括起始化合物和dmc催化剂的混合物。随后,允许进行与dmc催化剂的反应(例如,历时小于30分钟)。这允许使用dmc催化剂(例如,而不使用任何金属基路易斯酸,使得仅dmc催化剂用于形成烷氧基化的中间产物)制备烷氧基化的中间产物。随后,在不同于第一温度的第二温度下在具有活性的路易斯酸催化剂存在下,通过使用中间产物并且通过另外添加环氧丙烷、环氧乙烷和环氧丁烷中的至少一种到反应混合物来进行剩余物的聚合反应(例如,历时小于四十五分钟)。
使用环氧烷如环氧丙烷、环氧乙烷或环氧丁烷形成起始剂化合物自身。起始剂化合物可为二醇或三醇。举例来说,起始剂化合物为羟基官能基当量小于500g/mol当量的全部环氧丙烷基二醇或三醇。另外,含羟基引发剂化合物与环氧烷一起使用以形成起始剂化合物。含羟基引发剂化合物为在聚合反应中待烷氧基化的任何有机化合物。其含有1个或更多个羟基。其可含有多达12个或更多个羟基。可使用引发剂化合物的混合物。引发剂化合物将具有比聚醚产物的羟基当量小的羟基当量,例如,可具有30到500的羟基当量。例示性引发剂化合物包括(但不限于)乙二醇、二乙二醇、三乙二醇、丙二醇、二丙二醇、三丙二醇、1,4-丁二醇、1,6-己二醇、1,8-辛二醇、环己烷二甲醇、丙三醇、三羟甲基丙烷、三羟甲基乙烷、季戊四醇、山梨醇和蔗糖以及具有比聚合反应的产物的羟基当量小的羟基当量(例如,高达500g/mol当量)的这些中的任一种的烷氧基化物(尤其乙氧基化物和/或丙氧基化物)。
当路易斯酸催化剂添加到已经在dmc催化剂存在下进行烷氧基化方法的反应混合物时,反应器的温度相比于添加dmc催化剂时可减少至少20℃。根据例示性实施例,当dmc催化剂活化(例如,在环氧丙烷进料逐渐/缓慢添加到反应器的时间期间和在起始剂化合物与dmc催化剂混合的时间之后)时,反应器的第一温度(在分批或连续法中)可为125℃到160℃。可在未添加环氧烷进料且在添加路易斯酸之前的情况下,在允许进行形成中间产物的时间期间首先降低反应器的温度。当引入路易斯酸时,反应器温度可处于25℃到115℃和/或60℃到115℃的第二温度。在例示性实施例中,对含有活性dmc催化剂和活性路易斯酸的混合物的相对贡献的控制可使得路易斯酸能够支配在链端上添加环氧乙烷。
在例示性实施例中,当聚醚多元醇衍生自环氧丙烷基起始剂化合物(例如,聚环氧丙烷起始剂化合物)时,在第一温度下将环氧丙烷添加到混合物,并且在第二温度下将环氧丙烷、环氧乙烷或环氧丁烷添加到反应混合物。
聚醚多元醇可以两步骤、一锅式方法制备,所述方法以可使用dmc催化剂快速建构多元醇链的此类方式使用dmc催化剂和三(五氟苯基)硼烷,并且通过随后阶段添加三(五氟苯基)硼烷可在链端处产生伯羟基可。
聚合反应可在任何类型的适合于所遇压力和温度的容器中进行。例示性半分批法示出在图1中。例示性连续法使出在图2中。在连续或半连续法中,容器可具有一个或多个入口,在反应期间可经由所述入口引入环氧烷和额外引发剂化合物。在连续法中,反应器容器应当含有至少一个出口,局部聚合反应混合物的部分可通过所述出口抽出。具有单个或多个用于注入起始材料的位置的管状反应器、环流反应器和连续搅拌槽反应器(ctsr)是适合于连续或半连续操作的所有类型容器。例示性方法在美国专利公开案第2011/0105802号中论述。
在前述双催化剂系统基方法中的任一种方法中获得的所得聚醚多元醇产物可进一步处理,例如以闪蒸方法和/或汽提方法。举例来说,可处理聚醚多元醇以减少催化剂残留物,即使所述催化剂残留物可保持在产物中。通过对所述多元醇进行汽提可去除水分。根据实施例,聚环氧丙烷多元醇可具有15ppm到100ppm(例如,35ppm到100ppm,50ppm到75ppm等)的dmc浓度(在最终聚环氧丙烷多元醇中以ppm为单位)。根据实施例,聚环氧丙烷多元醇可具有100ppm至500ppm(例如,100ppm到250ppm)的路易斯酸催化剂浓度(在最终聚环氧丙烷多元醇中以ppm为单位)。
聚合反应可表征为“构造比率”,其被定义为聚醚产物的数均分子量与引发剂化合物的数均分子量的比率。所述构造比率可高达160,但更为通常在2.5到约65的范围内并且更为通常在2.5到约50的范围内。当聚醚产物具有85到400的羟基当量时,构造比率通常在约2.5到约15或约7到约11的范围内。
实施例涉及用于高伯羟基含量(例如,至少60%和/或约70%)和高分子量多元醇(例如,聚环氧丙烷多元醇)的催化方法。在例示性实施例中,所述一锅式方法涉及以顺序方式使用dmc催化剂和三(五氟苯基)硼烷(fab)。具体来说,通过分别在大于130℃和小于110℃的温度下执行dmc催化的反应和fab催化的反应,fab催化剂可用于在dmc催化剂存在下区域选择性地形成伯羟基。所述方法可用于从低分子量起始剂快速地合成高分子量产物。
根据双催化剂系统方法制备的聚醚多元醇可用于制备聚氨基甲酸酯。较高当量聚醚多元醇产物可用于制备弹性或半弹性聚氨基甲酸酯产物,包括非多孔或微孔弹性体、涂料、粘合剂、密封剂和柔性的、刚性及粘弹性聚氨基甲酸酯泡沫。柔性聚氨基甲酸酯泡沫可在板材或模制方法中制造。
除非另外指明,否则所有份数和百分比都是以重量计。
实例
分析方法:
凝胶渗透色谱法(gpc):用于测定数均分子量的gpc分析在1.0ml/min流动速率下使用4个串联连接的plgel有机gpc色谱柱(3μm安捷伦公司())和作为洗脱剂的四氢呋喃而进行。色谱柱温度为40℃。voranoltmcp6001、voranoltm210、230-660和230-056n用作内标。
伯羟基和仲羟基的测定(选择性):通过三氟乙酰化,随后19f-nmr分析来测定开环的选择性。使用在astmd4273-94中描述的步骤进行样本制备,并且对于用于聚醚多元醇的19f-nmr的样本制备sop可在弗里波特(freeport)分析科学-摩尔结构资料库中获得。如在astm方法中所陈述的,衍生化需要了解oh#或mw和多元醇的官能度,因为其决定用于多元醇衍生化的tfaa量。有必要添加足够量的tfaa以确保完成衍生化反应。
oh#可被计算为=33×%oh
%oh=1700/多元醇的羟基当量
多元醇的羟基当量=多元醇的mw/官能度基于羟基数,astm方法给出以下用于待添加到反应的tfaa量的建议。
19fnmr分析:使用brukeravanceiii400mhz光谱仪获取19fnmr波谱。使用64个瞬变扫描/数据文件,3秒脉冲重复延迟,光谱宽度93,750hz,和13k数据点的文件大小获取数据。使用饱和度恢复实验充分验证弛豫延迟。使用在cdcl3中处于0.1wt%的三氟-甲苯为内部化学位移标准获取波谱。
主要使用以下材料:
起始剂化合物1具有约700g/mol数均分子量的聚环氧丙三醇,即,低分子量po三醇(可voranoltm270够自陶氏化学公司())。
起始剂化合物2具有约450g/mol数均分子量的聚环氧丙三醇(可voranoltmcp450够自陶氏化学公司)。
起始剂化合物3具有约400g/mol数均分子量的聚环氧丙烷二醇(可voranoltmp400够自陶氏化学公司)。
dmc催化剂六氰钴酸锌催化剂络合物(以名称
fab三(五氟苯基)硼烷(购自博尔德科学())。
添加物包括磷酸的酸化剂。
根据下表1概括的条件,使用上述材料制备工作实例1到工作实例6和比较实例a到比较实例e。参考表1,如上文所论述,通过凝胶渗透色谱法测定数均分子量。如上文所论述,通过使用三氟乙酸酐的衍生化,随后进行19f-nmr)测定伯羟基含量(即,伯oh)。
表1
工作实例1为聚环氧丙三醇,其使用起始剂化合物1(即,具有约700g/mol的分子量的丙氧基化三醇)和顺序双催化剂方法制备,其中用于fab添加的第二温度比用于添加dmc的第一温度小20°。具体来说,工作实例1使用以下方法制备:500ml压力反应器装入起始剂化合物1(50g),添加剂(1.3μl的0.15m溶液)和dmc催化剂(0.024g)。通过在氮气充气下将混合物加热到130℃持续2小时来干燥。在阻断氮气流并封闭排气孔后,将环氧丙烷作为po进料缓慢地添加到反应器。dmc催化剂在约20至30分钟内活化,在此期间po进料逐渐提高到2.0ml/min至2.5ml/min。在使用po进料添加约240ml的po后,阻断进料并且使反应继续15分钟并冷却到60℃。其后,fab(0.080g)作为一部分添加并且将反应器加热到110℃。以约0.3ml/min至0.6ml/min的速率重新开始po进料。在添加约91ml的po后,使反应消化30分钟,并且用氮吹扫45分钟。
举例来说,关于工作实例1,可进行以下反应:
工作实例2为使用起始剂化合物1和顺序双催化剂方法制备的聚环氧丙三醇,其中用于添加fab的第二温度比用于添加dmc的第一温度小70°。具体来说,使用如上文关于工作实例1所论述的相同方法制备工作实例2,不同在于,在添加fab之后反应器保持在60℃。
比较实例a为使用起始剂化合物1和顺序双催化剂方法制备的聚环氧丙三醇,但用于添加fab的第二温度比用于添加dmc的第一温度高。具体来说,比较实例b使用以下方法制备:500ml压力反应器装入起始剂化合物1(50g)、添加剂(1.3μl的0.15m溶液)和dmc催化剂(0.024g)。通过在氮气充气下将混合物加热到130℃持续2小时来干燥。在阻断氮气流并且封闭排气孔后,将环氧丙烷作为po进料缓慢地添加到反应器直至压力达到20psi。随后,切断po进料并且使反应继续直至11分钟的时段并且压力达到8.9psi。此时认为催化剂“活化”并且重新开始po进料并逐渐增加到2.0ml/min至2.5ml/min。在使用po进料添加约330ml的po后,阻断进料并且使反应继续30分钟并在130℃下在用氮吹扫额外30分钟。随后,反应器冷却到70℃并且fab(0.080g)作为一部分添加,并且反应器搅拌30分钟以均匀化催化剂。
比较实例b为使用起始剂化合物1和顺序双催化剂方法制备的聚环氧丙三醇,但用于添加fab的第二温度比用于第一次添加的温度(140℃)高10℃。500ml压力反应器装入起始剂化合物1(50g)、磷酸(1.3μl的0.15m溶液)和dmc催化剂(0.024g)。通过在氮气充气下将混合物加热到130℃持续2小时来干燥。在阻断氮气流并且封闭排气孔后,将环氧丙烷缓慢地添加到反应器。dmc催化剂在约20至30分钟内活化,在这后,po进料逐渐增加到2.0ml/min至2.5ml/min。在添加239.4ml的po后,阻断进料并且使反应继续15分钟。随后反应器冷却到60℃并且一次性添加fab(0.080g),并将反应器加热到140℃。以0.3ml/min至0.6ml/min的速率重新开始环氧丙烷进料。在添加90.8ml的额外po后,使反应继续30分钟并且用氮气吹扫45分钟。
比较实例c为使用起始剂化合物1和顺序双催化剂方法制备的聚环氧丙三醇,但用于添加fab的第二温度与用于添加dmc的第一温度相同。具体来说,使用类似于如以上关于工作实例1所论述的方法制备比较实例c,不同在于在添加fab之后反应器被设定为130℃。
比较实例d为使用起始剂化合物2(即,具有约450g/mol的分子量的丙氧基化三醇)和dmc催化剂制备的聚环氧丙三醇,所述三醇未使用fab制备并且其具有相对低的伯羟基含量。具体来说,使用类似于以上关于比较实例a所论述的方法制备比较实例d,不同在于,使用833克的起始剂化合物2来代替起始剂化合物1(以及0.083克的添加剂和225mg的dmc催化剂)。另外,将反应器加热到140℃并且经受真空1小时。添加由环氧丙烷和环氧乙烷(按重量计98/2)组成的混合氧化物进料(92.6g)。使反应在相同温度下继续30分钟。重新开始混合氧化物进料(5010g,按重量计98/2的环氧丙烷/环氧乙烷),在这之后,添加由按重量计25/75的环氧丙烷和环氧乙烷组成的混合进料(1565g)。在30分钟之后,执行短时间的真空汽提。
使用比较实例d(即,使用起始剂化合物2制备的聚环氧丙三醇)制备工作实例3,其中随后在第二温度下添加fab,所述第二温度比用于添加dmc的第一温度小40°。使用类似于以上关于工作实例1所论述的方法进行fab的添加,不同在于第二温度被设定为90℃。具体来说,对于工作实例4,使用加压的不锈钢高压容器,向1l压力反应器中装入来自比较实例d的产物(约373克)和fab(0.113g)。在氮气充气下将反应器加热到90℃。在阻断氮气流后并封闭排气孔,以0.6ml/min将环氧丙烷(76.9ml)添加到反应器。使反应继续30分钟,用氮吹扫,并取样(1.5g)。在阻断氮气流并封闭排气孔后,使用质量流量控制器以0.35g/min的速率添加环氧乙烷(16g)。使反应继续30分钟并且用氮气吹扫45分钟。
工作实例4为使用起始剂化合物1和顺序双催化剂方法制备的聚环氧丙三醇,其中用于添加fab的第二温度比用于添加dmc的第一温度小50°,并且使用的dmc催化剂和fab催化剂两者的量均减少。具体来说,使用类似于以上关于工作实例1所论述的方法制备工作实例4,不同在于减少催化剂的量和第二温度被设定为90℃。具体来说,向500ml压力反应器中装入起始剂化合物1(50g)、添加剂(1.5μl的0.15m溶液)和dmc催化剂(0.01g)。通过在氮气充气下将混合物加热到140℃持续2小时来干燥。在阻断氮气流并封闭排气孔后,将环氧丙烷作为po进料以0.6ml/min添加到反应器。po进料逐渐增加到2.5ml/min并且在添加239.2mlpo的过程中反应器中的压力低于12psig。随后,阻断进料并且使反应继续15分钟并在氮气吹扫下冷却到90℃。然后,一次性添加fab(0.048g)并且以0.5ml/min的速率重新开始po进料。在添加84.7ml的环氧丙烷后,使反应继续30分钟,用氮气吹扫30分钟,并且冷却。
工作实例5为使用起始剂化合物3(即,具有约400g/mol的分子量的丙氧基化二醇)和顺序双催化剂方法制备的聚环氧丙烷二醇,其中用于添加fab的第二温度比用于添加dmc的第一温度小40°。具体来说,使用类似于以上关于工作实例1所论述的方法制备工作实例5,不同在于,使用起始剂化合物3来代替起始剂化合物1并且第二温度被设定为90℃。具体来说,向500ml压力反应器中装入起始剂化合物3(65g)、添加剂(2.3μl的0.15m溶液)和dmc催化剂(0.028g)。通过在氮气充气下将混合物加热到130℃持续2小时来干燥。在阻断氮气流并封闭排气孔后,将环氧丙烷作为po进料缓慢地添加到反应器。po进料在40分钟内逐渐增加到1.5ml/min至2.0ml/min并且在反应器中的压力在添加236.3ml的po过程中低于15psig。阻断进料并且使反应继续10分钟。氮气添加到反应器并且使内容物冷却。然后,一次性添加fab(0.095g)并且在氮气吹扫下将反应器加热到90℃。在90℃以0.5ml/min的速率重新开始po进料。在添加157.5ml的po后,使反应在90℃继续33分钟并且在90℃下用氮气吹扫40分钟。
比较实例e为使用起始剂化合物3和dmc催化剂制备的聚环氧丙烷二醇,所述二醇未使用fab制备并且其具有相对低的伯羟基含量。具体来说,使用类似于以上关于比较实例a种所论述的方法制备比较实例e为,不同在于,使用起始剂化合物3来代替起始剂化合物1并且第一温度被设定成140℃。具体来说,向1l压力反应器中装入起始剂化合物3(125g)、添加剂(1.85μl的0.15m溶液)和dmc催化剂(0.03g)。通过在氮气充气下将混合物加热到140℃持续2小时来干燥。在阻断氮气流并封闭排气孔后,将环氧丙烷作为po进料以1.0ml/min添加到反应器。po进料逐渐增加到3.5ml/min并且反应器中的压力在添加132.2mlpo的过程中低于9psig。然后,阻断进料并且使反应继续8分钟。随后,以0.6ml/min的进料速率将环氧丁烷(bo)添加到反应器,所述速率在35分钟内增加到3.5ml/min。反应器中的压力在408.7ml的bo添加过程中低于17psig。然后,阻断进料并且使反应消化15分钟并冷却到70℃。
使用比较实例e(即,使用起始剂化合物3制备的聚环氧丙烷二醇)制备工作实例6,其中随后在第二温度下添加fab,所述第二温度比用于添加dmc的第一温度小50°。使用类似于以上关于工作实例1所论述的方法进行fab的添加,不同在于第二温度被设定为90℃。具体来说,将fab(0.108g)一次性添加到比较实例e的样品中。圆底烧瓶转移到旋转式蒸发器并且在85mbar的真空和氮气吹扫下加热到110℃,维持20分钟。使用氮气使烧瓶的内容物达到1大气压,并且在内容物是热的时将其经由漏斗转移到压力反应器中。在氮气吹扫下将压力反应器加热到90℃,然后停止吹扫并且封闭排气孔。随后,以0.5ml/min的速率重新开始bo进料。在添加约202.7ml的bo后,使反应消化30分钟,并且用氮吹扫45分钟,并且冷却。
- 还没有人留言评论。精彩留言会获得点赞!